Master Qualité - Communication
publique des résultats d'un stage de fin d'études Master Qualité
- UTC - rue du docteur Schweitzer - CS 60319 - 60203
COMPIEGNE Cedex - France - master-qualite@utc.fr
- Téll : +33 (0)3 44 23 44 23
Avertissement
: Si vous arrivez directement sur cette page, sachez
que ce travail est un rapport d'étudiants et doit être
pris comme tel. Il peut donc comporter des
imperfections ou des imprécisions que le lecteur doit
admettre et donc supporter. Il a été réalisé pendant
la période de formation et constitue avant-tout un
travail de compilation bibliographique, d'initiation
et d'analyse sur des thématiques associées aux
concepts, méthodes, outils et expériences sur les
démarches qualité dans les organisations. Nous ne
faisons aucun usage commercial et la duplication est
libre. Si, malgré nos
précautions, vous avez des raisons de contester ce
droit d'usage, merci de nous
en faire part, nous nous efforcerons d'y
apporter une réponse rapide. L'objectif de la
présentation sur le Web est de permettre l'accès à
l'information et d'augmenter ainsi les échanges
professionnels. En cas d'usage du document, n'oubliez
pas de le citer comme source bibliographique.
Bonne lecture...
"Vali-Médic'" :
une aide à la démarche de validation des méthodes
analytiques
Référence
bibliographique à rappeler pour tout usage :
« VALI- MEDIC’ » : une aide à la démarche de validation de méthodes analytiques, Marlène FERDERIN, Université de
Technologie de Compiègne, Master Qualité et
Performance dans les Organisations (QPO), Mémoire d'Intelligence Méthodologique du stage
professionnel de fin d'études, juin 2017, www.utc.fr/master-qualite,
puis "Travaux", "Qualité-Management", réf n° 391, https://doi.org/10.34746/fna7-sf69
RÉSUMÉ
Le secteur
de l’industrie pharmaceutique est un secteur très
concurrentiel et en constante expansion. Avec
l’émergence des startups et des biotechnologies les
clients et patients sont de plus en plus demandeurs de
nouveaux traitements. C'est pourquoi, dans les années
80, les autorités pharmaceutiques ont mis en place des
réglementations strictes afin d’assurer la sécurité des
médicaments dans l'ensemble du processus de
développement et de production. La commercialisation
d’un médicament nécessite la rédaction de rapports de
validation des méthodes analytiques garantissant la
qualité et la fiabilité des médicaments produits.
L’approche « Vali-Medic’ » à pour objectif d'aider les
ingénieurs « Validation de méthodes analytiques (VMA ) »
à réussir cette étape de validation cruciale pour la
pérennité, la compétence et la compétitivité des
laboratoires pharmaceutiques.
Mots clés : Validation des
méthodes analytiques, industries pharmaceutiques,
production
ABSTRACT
The pharmaceutical industry is a very
competitive and growing sector. Following the emergence
of start-ups and biotechnology customers and patients
are enquiring new and more effective treatments. This is
why, the pharmaceutical authorities established strict
regulations to ensure the safety of drugs throughout
their development and production process. The marketing
of a medicinal product requires the writting of
validation reports on analytical procedures to guarantee
the quality and reliability of medicines produced. The
"Vali-Medic" approach aims to help "Validation of
Analytical Procedures (VMA)" engineers to pass this
crucial validation step for the sustainability,
competence and competitiveness of pharmaceutical
laboratories.
Keywords: pharmaceutical
industry, validation of analytical procedures,
production
L'industrie pharmaceutique appartient au
secteur économique regroupant les activités de recherche et
développement (R&D), de fabrication et de commercialisation
des médicaments à usage humain ou vétérinaire. C’est l’un des plus
important au niveau mondial. Se retrouvent dans l’industrie
pharmaceutique : les laboratoires pharmaceutiques (Big Pharma) et
les laboratoires de biotechnologie qui sont en perpétuelles
innovation via le développement de nouvelles de techniques,
technologies et médicaments.
La conception d’un médicament est une processus
long (15 ans en moyenne) et complexe. En effet, avant d’arriver à
sa forme définitive, le médicament passe par plusieurs phases de
R&D et de production. Afin que le médicament puisse être
commercialisé, un dossier d’autorisation de mise sur le marché
(AMM) doit être réalisé. Pour cela une réglementation stricte a
été mise en place afin de garantir l’efficacité et la sécurité du
médicament sur l’Homme ou l’animal. C’est à cette étape
qu’intervient la validation des méthodes analytiques. En effet, il
est essentiel que chaque tests et expérimentations réalisés sur la
molécule médicament soient 100% fiables.
Selon une étude menée par la Leem en 2015 [1],
la France a investi en 2013 plus de 4.000 M€ en recherche et
développement, c’est-à-dire un investissement d’environ 10% du
chiffre d’affaires d’un laboratoire. En 2014, en moyenne un
Français achetait pour 516€ de médicaments par an. Par ailleurs,
l’exportation et l’importation de médicaments en France joue un
rôle important dans l’économie de l’industrie pharmaceutique
puisqu’environ 7 500 M€ d’excédent commerciaux sont dégagés par
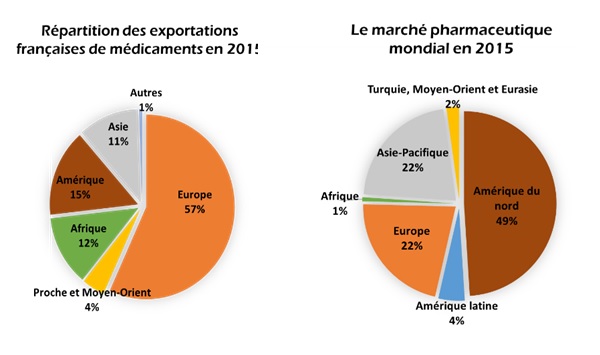
les médicaments. En effet, la France exporte essentiellement vers
l’Europe mais aussi en Amérique et en Afrique (Figure 1.a).
Dans le marché mondial de l’industrie, la France possède une part
de 3.5% et se retrouve 5ème du classement après les États-Unis, la
Chine, le Japon et l’Allemagne. En 2015, L’Europe et l’Asie
possèdent des parts de marché équivalentes se plaçant juste après
l’Amérique du nord qui domine le marché de l’industrie
pharmaceutique (Figure 1.b) [2].
Figure 1: L'industrie pharmaceutique en chiffres [source : auteure]
Il existe différentes catégories de
laboratoire pharmaceutiques :
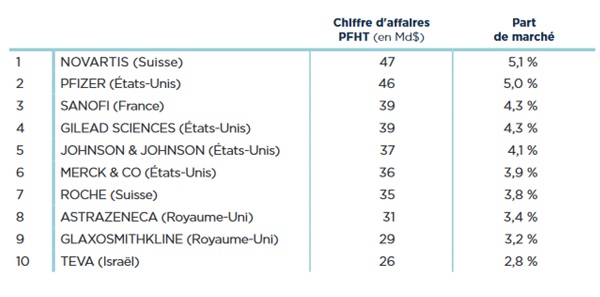
Les Big Pharma c’est-à-dire les leaders du marché
pharmaceutique mondial. Ils réalisent en général la plupart
du travail dans la conception d’un médicament (R&D,
Production et Commercialisation) (Figure 2)
Les laboratoires de biotechnologie, qui
généralement ne réalisent qu’une partie du travail
d’élaboration d’un médicament (R&D essentiellement) et
ont parfois recours à des prestataires de services afin de
réaliser des tests spécifiques qu’ils ne peuvent faire
eux-mêmes.
Les start-ups de biotechnologie, en pleine
expansion sur le marché pharmaceutique arrivent avec des
idées nouvelles et des technologies innovantes.
Les laboratoires de service (prestataires) vendent
leurs services aux autres industries qu’ils soient Big
Pharma ou start-ups. Il existe 2 types de prestataires
: les CRO (Contract Research Organization) et CMO
(Contract Manufacturing Organization). Les CRO permettent
aux entreprises de sous-traiter des tests de R&D qu’ils
ne peuvent réaliser alors que les CMO permettent de
sous-traiter la production du médicament mais également
quelques parties de R&D notamment lors des tests en
phase clinique.
Les laboratoires d’analyses médicales ou
laboratoires de biologie médicale, réalisent des analyses de
différents fluides biologiques (humains ou animales) pour le
diagnostic de possibles maladies.
Figure 2 : Les principales Big
Pharma mondiales en 2015 [source : [2]]
1.2.
Le rôle de la validation de méthodes analytiques dans la
conception d’un médicament
1.2.1. Les phases de développement d’un
médicament
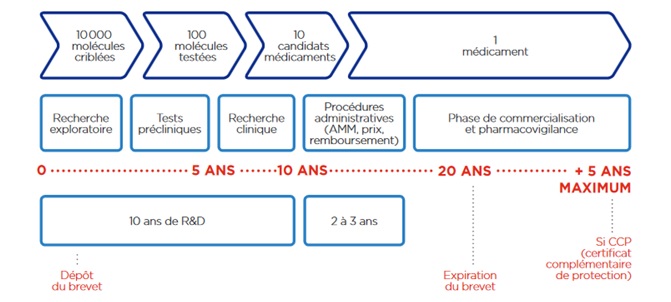
Le processus de fabrication et de
commercialisation d’un médicament est un procédé long et coûteux
à mettre en place. En effet, il faut compter entre 15 et 20 ans
de R&D avant de commercialiser le médicament.
Les principales étapes pour la réalisation d’un médicament sont
(Figure 3) :
La recherche exploratoire, où des milliers de
molécules jouant potentiellement un rôle dans la pathologie
ciblés sont testées. Un criblage à haut débit est réalisé
afin de diminuer le nombre de candidat médicament.
Les tests précliniques ou non cliniques, consistent à
réaliser des tests cellulaires et des tests sur animaux.
Les essais cliniques, se caractérisent en 4 phases [3]:
o Phase I : La première
administration de la molécule à l’homme (volontaire sain)
afin de réaliser une évaluation préliminaire du médicament à
la dose déterminée en phase préclinique et de vérifier sa
tolérance par l’Homme. Seul un petit nombre de volontaire
sont testés (10 à 40).
o Phase II : Cette
étape permet de confirmer l’efficacité et la tolérance du
médicament sur l’homme et à déterminer la posologie optimale
pour les malades. Lors de cette phase des études de
pharmacocinétiques humaines et d’interactions
médicamenteuses peuvent être réalisées. Quelques centaines
de malades sont testés.
o Phase III : Ces
tests sont réalisés sur des milliers de patients afin de
représenter une population variée de la maladie ciblée. Ces
tests réalisés en double aveugle (le médecin et le patient
ne savent pas quelle molécule est prescrite) permet de
comparer le nouveau médicament avec un standard et un
placebo afin de déterminer son efficacité. Ainsi, l’intérêt
thérapeutique de la molécule médicament peut être démontré
et le rapport bénéfices/risque évalué. A cette étape le
médicament est sous sa forme définitive et un nombre
important de contrôle de sécurité, de validation, etc. ont
été réalisés. A la suite de cette étape (Phase III) le
dossier d’AMM peut être rédigé et soumis à l’ANSM (Agence
Nationale de Sécurité du Médicament).
o Phase IV : Cette
phase appelée pharmacovigilance est réalisée après la
commercialisation du médicament sur un nombre très important
de patients. Cette étape permet de connaitre les effets à
plus long terme du médicament et de connaitre ses effets à
grande échelle.
1.2.2. La validation de méthodes analytiques
(VMA)
Avant sa
commercialisation un médicament passe par différentes phases
cruciales où de nombreux tests sont réalisés. Il est essentiel que
les résultats de ces tests soient fiables et que toutes les
opérations réalisées soient tracées que ce soit lors de la R&D
ou lors de la production du médicament. En effet, s’il y a eu un
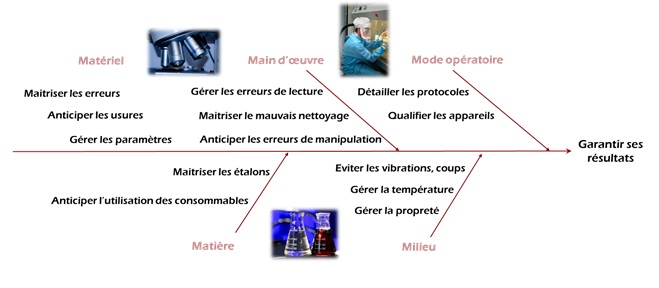
changement, il est important que celui-ci soit consigné. Lors des
tests analytiques, des incertitudes dans la mesure peuvent
facilement être induite. Par exemple, il est possible qu’il y ait
eu des erreurs de manipulation ou encore des perturbations du
signal (Figure 4). C’est pourquoi il est primordial de tracer et
d’avoir recours à la validation des méthodes analytiques afin de
paramétrer tous les facteurs potentiellement perturbateurs du
signal et de réaliser de la même manière toutes les
expérimentations.
Le but de la
validation est de démontrer que la méthode d'analyse est adaptée à
l'usage auquel elle est destinée. Il est obligatoire d’utiliser
des standards et des contrôles bien caractérisés (avec une pureté
connue) lors de toutes les étapes de validation.. Elle est
réalisée après les étapes de R&D et de mise au point du
principe actif. Elle s’applique pour les procédures d’analyses des
matière premières, la formulation galénique du médicament, tous
les produits de production (intermédiaires et finaux) et les
essais de stabilité.
Figure 4 : Pourquoi la validation
des méthodes analytiques permet de garantir ses résultats [source : auteure]
Valider des méthodes analytiques
lors de la validation pharmacologique et pharmaceutique d’une
molécule ou d’un médicament est obligatoire pour un laboratoire.
Par exemple, l’activité des CRO et CMO est en partie basée sur la
vente des services qu’ils proposent c’est-à-dire des services
respectant les réglementations en vigueurs. Si ces laboratoires
n’appliquaient ni BPL ni BPF, ils n’auraient aucun client.
Puisque, la validation des méthodes analytiques est obligatoire
lors de la conception d’un médicament, l’enjeu majeur de la mise
en place de la VMA est la pérennité du laboratoire. Mettre en
place une démarche de VMA peut sembler contraignante, mais cette
démarche doit être adaptée au fonctionnement du laboratoire et à
ses objectifs. La VMA est un processus progressif passant par
plusieurs étapes dès le développement de la méthode. Un fois
développée la méthode subit une étape de pré-validation puis que
qualification. Ensuite, la validation est réalisée suivi par la
rédaction d'un rapport de validation conformément aux
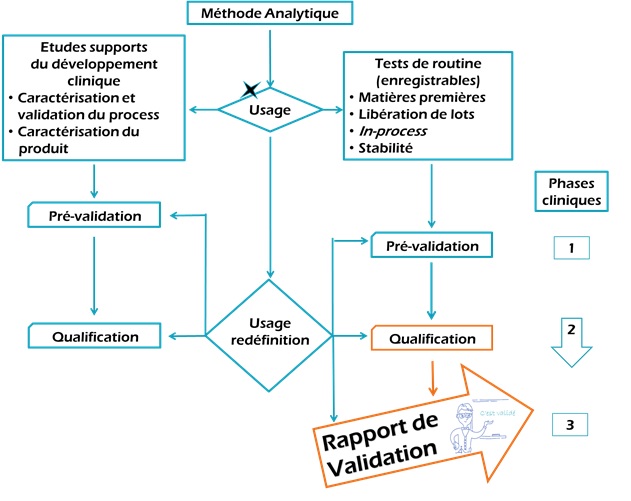
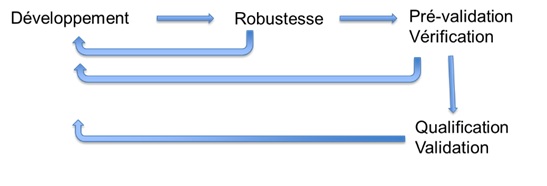
règlementations en vigueur (Figure 5).
Figure 5: Validation en fonction
de la méthode analytique et de la phase clinique [source : auteure]
1.3.
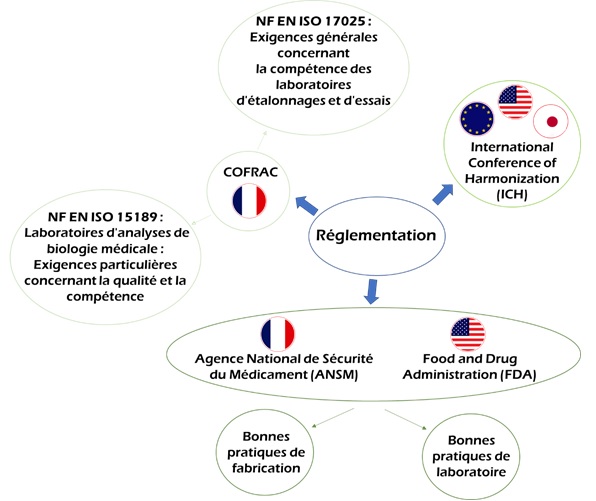
Les réglementations en vigueur concernant la VMA
Afin d’assurer la fiabilité des résultats des
analyses réalisées, certains organismes ont mis en place une
réglementation stricte. En effet, afin de pouvoir commercialiser
les médicaments sur le marché Français ou mondial, les entreprises
doivent soumettre un dosser d’AMM à l’ANSM (France) où tous ces
renseignements sont précisés. De plus, l’ANSM [4], agence
d’expertise contribuant à élaborer les règlementations
pharmaceutiques françaises et à contrôler leur application dans
les laboratoire accrédités, demande pour chaque molécule
médicament soumise à la commercialisation, que certains tests
soient réalisés dans les respects de bonnes pratiques de
laboratoire et de fabrication (BPL et BPF). Aux Etats-Unis, la FDA
(Food and Drug Administration) [5] a même élaboré un texte de loi
obligatoire, les cGMP (current Good Manufacturing Practices), que
chaque organisme de production voulant travailler aux ou avec les
USA doit respecter, ce texte de loi est très proche des BPF que
l’on retrouve en France. Pour la pérennité des laboratoires, il
est primordiale de réaliser les tests et la production d’un
médicament conformément aux BPF et BPF.
Par ailleurs, l’ICH
(International Conference of Harmonization), un comité d’expert
regroupant les autorités de réglementation ainsi que les leaders
mondiaux de l’industrie pharmaceutique, c’est-à-dire les
Etats-Unis, l’Europe et le Japon a mis en place un guide
(Validation et procédure analytique : texte et méthodologie)
[6]. Le respect de ce guide est primordial pour la
commercialisation du médicament. L’ICH a pour but d’harmoniser
mondialement les affaires de santé afin d’assurer la sécurité,
l’efficacité et la qualité des médicaments produits.
De plus, la pharmacopée (européenne, américaine, japonaise,
française, etc.), un ouvrage réglementaire définissant les
critères de pureté des matière premiers ou des préparations
médicamenteuses mais aussi les méthodes d’analyses à mettre en
place assurant ainsi le contrôle des médicaments produits. La
description de ces méthodes sert d’aide pour la mise en place du
plan de validation de la VMA [7].
Figure 6: La réglementation en
vigueur concernant la validation des méthodes analytiques [source : auteure]
La VMA est donc un point clé dans le
développement et la pérennité d’un laboratoire. C’est grâce à
celle-ci et aux règlementations respectées par le laboratoire que
celui-ci a la possibilité de séduire de nouveaux clients tout en
fidélisant les anciens. En effet, afin de commercialiser un
médicament il est obligatoire que les VMA soient respectées à
chaque phase du développement du médicament. Il est donc possible
de se demander « Comment mettre en place une démarche de VMA
efficace et simple au sein d’un laboratoire ? »..
«
Vali-Médic ’ » est un projet simple à mettre en place par les
ingénieurs et les techniciens « VMA » et adapté à toutes les
entreprises. « Vali-Médic’ » est un guide visant à aider les
entreprises dans leur organisation, en impliquant le personnel
dans le projet et le sensibilisant à l’importance de la validation
des méthodes analytiques. « Vali-Médic’ » est valable pour toutes
les phases de développement d’un médicament et peut donc être une
aide lors de la conception d’un plan de validation et de la
rédaction des rapports de validation des méthodes.
2.2.
Le PTDCA, une démarche organisationnelle et opérationnelle
des VMA
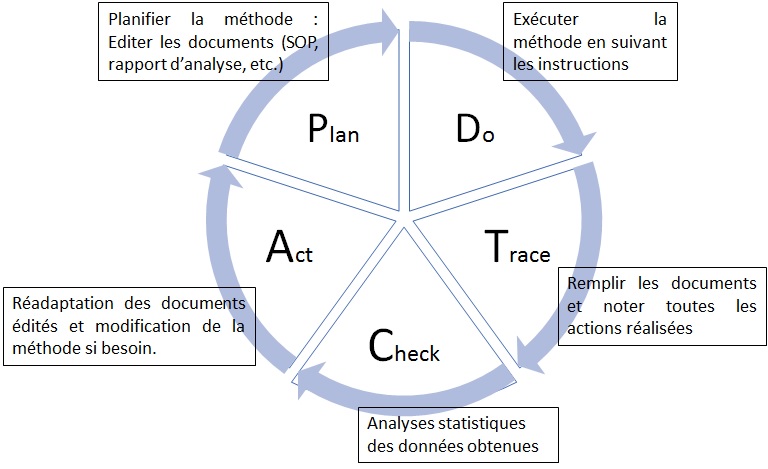
La démarche PDTCA (Plan, Do, Trace, Check, Act)
proposée est basée sur la méthode PDCA (Plan, Do, Check, Act)
adaptées à la validation des méthodes analytiques (Figure 7).
L’étape « Plan » consiste à
planifier et à prévoir la ou les actions à mettre en place en
prenant en comptes les différentes variables se présentant
(ressources, responsabilités et autorité, délais à respecter,
etc.). « Do » signifie « faire », à cette étape de la méthode, les
actions planifiées ultérieurement sont mises en place et réalisées
en respectant les instructions données. De plus, grâce à l’étape «
Trace » toutes les actions réalisées auront été notées et
répertoriées afin d’avoir une parfaite traçabilité et ainsi
pouvoir détecter les déviations rapidement lors de la phase «
Check ». Lors de cette phase, les résultats obtenus et les actions
réalisées et tracées sont vérifiés afin de détecter d’éventuelles
erreurs ou déviations par rapport au mode opératoire.
Les
différents résultats sont comparés et des analyses statistiques
sont réalisées afin de vérifier la concordance et la validité des
résultats selon la réglementation en vigueur [8]. Enfin lors de la
phase « Act », les actions à mettre en œuvre afin
d’adapter/réadapter ce qui a été réalisé précédemment sont
réfléchis, notées et réalisée. La boucle PDTCA recommence jusqu’à
ce qu’il n’y ait plus aucune modification de la méthode à
réaliser. En effet, une fois la méthode validée tout changement
dans le protocole doit également subir une étape de validation
afin de certifier que ce changement n’induit pas de modification
du résultat. Cette méthode propose de réaliser chaque étape de la
validation de méthodes analytiques en suivant un ordre en logique
afin de respecter et appliquer la réglementation en vigueur.
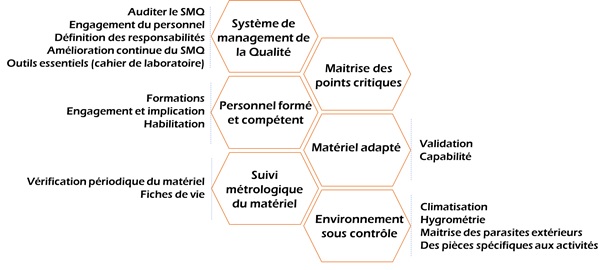
Afin que
« Vali-Médic ’ » soit correctement appliquée et corresponde
également à la démarche VMA, des points clés de réussites sont
proposés (Figure 8). En effet, pour maximiser ses chances de
réussite et être performant, « Vali-Médic’ » conseille de mettre
en place, un système de management de la Qualité qui va pouvoir
garantir les différents processus, approuver les documents édités
et sensibiliser le personnel à l’importance de la démarche.
Maitriser les points critiques, c’est-à-dire, les connaitre, les
définir, les anticiper et savoir les gérer afin de pouvoir
garantir l’efficacité de la méthode. Pour une VMA efficace, il est
primordial que le personnel soit formé, motivé et compétent pour
la mission proposée. Ensuite, afin que les VMA soient réalisées
dans de bonnes conditions il est essentiel d’avoir un matériel
adapté à la méthode, de réaliser un suivi métrologique de celui-ci
afin de garantir son bon état et de travailler dans un
environnement contrôlé, c’est-à-dire un environnement maitrisé et
où tous les paramètres sont connus afin que ceux-ci n’influencent
pas les résultats des tests.
Figure 8: "Vali-Médic'": les
points clés [source : auteure]
2.4.
Processus de validation de méthodes analytiques proposé
Le
processus de validation des méthodes analytiques suit le schéma
suivant. Il faut d’abord développer la méthode puis tester sa
robustesse, c’est-à-dire tester tous les points critiques et les
limites de la méthode pour finalement arriver à une phase de
pré-validation puis qualification et finalement de validation
(Figure 9).
Figure 9: Processus de validation des méthodes analytiques [source : auteure]
Le développement d’une méthode consiste à :
Choisir la méthode et l’équipement
Définir les étapes de préparation des échantillons
Choisir la gamme analytique
Vérifier spécificité / sélectivité
Il est important de vérifier la stabilité des solutions afin de :
Diminuer les sources de variation
Simplifier le test au maximum
Les tests de robustesse sur une
méthodes peuvent être réalisée lors de son développement. Elle
permet de mesurer sa capacité à ne pas être affectée par des
modifications faibles, délibérées ou encore par des facteurs
associés à la méthode. La robustesse donne également une
indication sur la fiabilité de la méthode dans les conditions
normales d’application. Lors des tests de robustesse, il ne faut
pas seulement tester les stabilités des solutions, il faut
également analyser les effets de variations délibérées de
paramètres clés sur les résultats. Si aucun effet est remarqué, il
faut préciser les variations possibles et s’il y a un effet
significatif, il faut signaler l’importance du paramètre et le
fixer afin d’assurer la robustesse de la méthode d’analyse.
La phase de pré-validation ou de vérification prend également part
au développement de la méthode d’analyse. A cette étape, il faut :
Atteindre un niveau d’exactitude de 100%, avec plusieurs
répliquats pour d’estimer la répétabilité
Confirmer des points de la méthode tels que la spécificité,
sélectivité, stabilité des solutions s’ils n’ont pas été
confirmé lors de son développement
Des résultats exacts et répétables
Atteindre le stade la
qualification d’une méthode d’analyse c’est presque atteindre le
stade de la VMA. En effet, la principale différence entre ces deux
étapes est la détermination des critères d’acceptabilité qui reste
optionnelle lors de la qualification mais qui est obligatoire en
validation.
La VMA a pour but de vérifier que la méthode possède
les performances demandées pour l’usage auquel elle est destinée
en considérant des limites d’acceptation prédéfinies ainsi que le
risque relatif à l’usage futur de la méthode. La VMA est un
processus progressif du développement d’un produit. La validation
complète n’est requise que lors de la réalisation du dossier
d’AMM. Un certain nombre de paramètres sont étudiés en fonction de
la nature de la méthode, de le l’usage à laquelle elle est
destinée et du stade de développement du produit (Figure 10).
Il
est intéressant de noter que les méthodes décrites dans les
Pharmacopées ne sont pas à revalider mais à vérifier dans certains
cas. « Lorsque les données présentées [..] ont été obtenues par
des méthodes de la pharmacopée, elles peuvent s'appuyer sur des
données de validation beaucoup plus sommaires, car il est admis
que les méthodes de la pharmacopée ont déjà été correctement
validées. » [9].
Les règles d’or de la validation des méthodes d’analyse sont les
suivantes :
Valider toute la méthode
Valider sur tout l’intervalle de dosage
Valider sur toutes les matrices/supports
Figure 10 : Degré de validation
en fonction du stade de développement d'un médicament [source : auteure]
QbD1
Phase
1
Phase
2et 3
Dossier
AMM
Contrôle
des matières premières
NA
Pre-validation
Qualification
Validation
Tests
libératoires pour les DS2 et les DP3
Pre-validation
Pre-validation
Qualification
Validation
Etudes
pré-cliniques/ toxicologies
NA
Pre-validation
Qualification
Validation
Etudesde
comparabilité/ caractérisation
NA
Qualification
Immunomonitoring
NA
Pre-validation
Qualification
Validation
Dosage
de bio-marqueurs
NA
Validation
1 QbD : Quality by Design
2 DS : Drug Substance (produit non formulé)
3 DP : Drug Product (produit formulé et réparti)
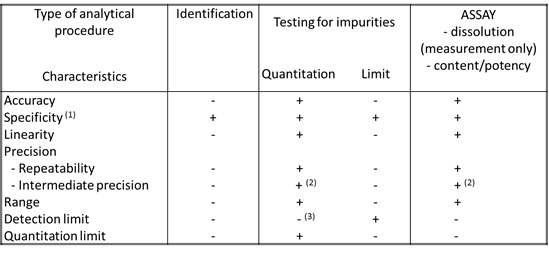
L’ICH a
établi un tableau regroupant les différents paramètres à valider
selon le types de méthodes analytiques (Figure 11). Ce tableau
permet de savoir quels sont les points importants à valider et
quels types de tests réaliser pour valider la méthode
analytique.
Figure 11: Paramètres de la validation pour les
différents types de méthodes analytiques [source : [5]]
(1) L’absence de «
specificity » d’une méthode analytique peut être compensée par
d’autres méthodes analytiques pour compléter le test
(2) Dans le cas où la «
reproducibility » a été réalisée, « l’intermediate precision »
n’est pas requise
(3) Peut être nécessaire dans
certains cas
Afin que
la création du dossier de validation et que toutes les étapes
citées précédemment ne soient pas trop longues et n’induisent pas
une perte de temps, il est intéressant de suivre les étapes
listées ci-dessous :
Décrire avec précision la méthode, c’est-à-dire donner tous
les détails de la méthode (environnement, le but, les
opérateurs, etc.) ;
Définir les différentes étapes afin de donner plus de clarté
à la méthode et de l’organiser plus facilement ;
Noter les matières, réactifs, appareillages critiques, etc.,
il est important de préciser le fournisseur, la référence, le
numéro de lot, les dates, etc. de chaque produit ou appareil ;
Expliciter les points à valider, c’est-à-dire donner les
point critiques et expliquer leur intérêt et leur importance
dans la méthode ;
Définir une stratégie de validation pour chaque point,
chaque point doit-être validé et se reporter à un contrôle ou
un étalon. Un trace écrite (manuscrite et/ou informatique) des
données est obligatoire ;
Définir un intervalle de confiance pour chaque paramètre. En
effet, pour prévenir de tout risque de déviation ou autres, il
faut que chaque paramètre du test appartienne à un intervalle
de confiance afin de réduire au maximum les risques de non
validation de la méthode ;
Définir une stratégie validant l’ensemble de la manipulation
;
Qualifier le matériel pour la méthode ;
Réaliser les manipulations selon la méthode d’analyse
décrite ;
Analyser les résultats et réaliser les statistiques (normes
ISO 5725) ;
Evaluer la robustesse du test, c’est-à-dire montrer la
fiabilité du test avec les écarts des paramètres déterminés
dans la méthode d’analyse ;
Réaliser les manipulations selon la méthode d’analyse
décrite ;
Analyser les résultats et réaliser les statistiques (normes
ISO 5725) ;
Écrire le dossier de validation explicitant tous les points
précédents et les données statistiques telles que : les
résultats de spécificité, linéarité, rang, fiabilité,
précision, les limites de détections et les limites de
quantification ;
Mettre en place un plan de validation de
méthode analytique et l’exécuter peut prendre entre 6 mois à 2 ans
selon la méthode. La validation de méthode est essentielle afin
d’assurer la sécurité et la fiabilité des médicaments produits.
Lors des phases cliniques, un grand panel de patients sont traités
avec le médicament, c’est pourquoi il faut que tout soit contrôlé
et qualifié afin que les patients puissent être traités en toute
sécurité même si le médicament n’est pas encore commercialisé et
validé via le dossier d’AMM.
Le guide « Vali-Médic’
» , téléchargeable gratuitement, est un document de vulgarisation
de la validation des méthodes analytiques pour une compréhension
simple et rapide.
Ce guide est destiné aux ingénieurs et
techniciens « VMA » pour permettre aux entreprises d’être
performantes tant au niveau fonctionnel qu’opérationnel. Il
détaille les étapes de la validation et ses points clés. Il aide
également à la rédaction d’un rapport de validation nécessaire au
dépôt du dossier d’AMM. « Vali-Médic’ » est proposé pour assurer
la pérennité des laboratoires pharmaceutiques.
A l’aide de «
Vali-Médic ’ » et de sa mise œuvres, les VMA seront réalisées tout
en respectant la règlementation en vigueur qu’elle soit française
ou internationale. Les entreprises pourront ainsi commencer la
production de leur médicament en toute sécurité et garantir aux
clients et patients la sécurité des médicaments produits. Les
tests validés pourront donc être appliqués en routine [11].
La
réglementation en vigueur dans l’industrie pharmaceutiques est
stricte et complexe afin de protéger aux maximum les patients mais
aussi les entreprises elles-mêmes. Le contrôle et la validation
des méthodes sont essentiels pour la commercialisation d’un
médicament.
Le guide VMA est donc un outil proposé pour aider les
ingénieurs et les techniciens à l’application de ces
réglementations. Il pourra également servir aux qualiticiens afin
de sensibilisé les équipes à l’importance et à l’intérêt du
respect de ces règlementations. En effet, il est essentiel que
tout le personnel se sente impliqué et concerné par l’application
de ces bonnes pratiques et normes.
[3] CSL Behring
France, « Phases d’étude clinique ». CSL Behring France,
janv-2014.
[4] « ANSM :
Agence nationale de sécurité du médicament et des produits de
santé ». [En ligne]. Disponible sur: http://ansm.sante.fr/.
[Consulté le: 06-juin-2017].
[5] « U.S. Food
& Drug Administration ». [En ligne]. Disponible sur:
https://www.fda.gov. [Consulté
le: 06-juin-2017].
[7] « Pharmacopée
française - 11ème édition ». Ed ANSM, Paris, http://ansm.sante.fr, 2017.
[8] «
International Council for Harmonisation (ICH) of Technical
Requirements for Pharmaceuticals for Human Use ». [En ligne].
Disponible sur: http://www.ich.org.
[Consulté le: 15-juin-2017].
[9] « Assurance de la qualité des produits
pharmaceutiques, Recueil des directives et autres documents ». Ed
OMS, Genève, www.who.int, 1998.
[11] M. Ferderin,
« « Vali-Médic’ » : une aide à la démarche de validation de
méthodes analytiques », Université de Technologie de Compiègne,
Master Qualité et Performance dans les Organisations (QPO),
Mémoire d’Intelligence Méthodologique du stage professionnel de
fin d’études,
www.utc.fr/master-qualite, puis « Travaux » «
Qualité-Management » réf n° 391, juin 2017.