|
Avertissement
|
|
Si vous arrivez
directement sur cette page, sachez que ce travail est un
rapport d'étudiants et doit être pris comme
tel. Il peut donc comporter des imperfections ou des
imprécisions que le lecteur doit admettre et donc
supporter. Il a été réalisé
pendant la période de formation et constitue
avant-tout un travail de compilation bibliographique,
d'initiation et d'analyse sur des thématiques
associées aux technologies biomédicales.
Nous
ne faisons aucun usage commercial et la duplication est
libre. Si vous avez des raisons de contester ce droit
d'usage, merci
de nous en faire part .
L'objectif de
la présentation sur le Web est de permettre
l'accès à l'information et d'augmenter ainsi
les échanges professionnels. En cas d'usage du
document, n'oubliez pas de le citer comme source
bibliographique. Bonne lecture...
|
Référence
à rappeler : Mise
en œuvre d'un banc de test de sécurité
électrique selon la norme ISO 17025, R. Gigleux, M.
Iracane, Projet DESS "TBH", UTC, 01-02
URL :
https://www.utc.fr/~farges/dess_tbh/01_02/Projets/acc_secu_elec/accreditation.htm
Mise en
oeuvre d'un banc de test de sécurité
électrique
selon la norme ISO
17025
|

Marion
IRACANE
|

Robin
GIGLEUX
|
REMERCIEMENTS
Nous remercions notre tuteur: M. FARGES Gilbert pour nous avoir
proposé ce sujet, pour son encadrement et ses conseils
avisés qui nous ont permis de mener à bien ce projet.
Ce sujet nous aura permis de démystifier
l'accréditation ISO 17 025 tout en nous permettant de
manipuler de nombreux outils de la qualité.
Nous tenons également à remercier le groupe
d'étudiants du module GE37 «Gestion de Projet »
(Cécile, Arnaud, Jonathan, Guillaume et Jérémie)
avec lequel nous avons collaborer, pour leur travail efficace, leur
sympathie et leur constante bonne humeur.
Nous remercions également M. Caliste pour ses ultimes conseils
sur le test de sécurité électrique
accrédité et M. Chevallier pour les divers moyens
techniques mis à notre disposition.
|
RESUME
L'accréditation
décernée par le COFRAC (comité
français pour l'accréditation), permet aux
laboratoires d'essais de prouver la qualité de leur
organisation et d'assurer la qualité de leurs
résultats.
Ce rapport aborde les différentes
étapes de la mise en place d'une démarche
qualité, au sein d'un laboratoire
réalisant des tests de sécurité
électrique sur des dispositifs
médicaux.
Dans une première partie, les
exigences de la norme ISO 17025 sont explicitées dans
le cadre de ce laboratoire. Dans une deuxième partie,
une méthodologie de mise en place est proposée
à l'aide d'une décomposition des
activités sous forme de processus. Enfin, certains
éléments sont explicités afin d'aider
à l'élaboration du manuel qualité, des
procédures, des instructions et les
enregistrements associés.
|
|
ABSTRACT
Accreditation awarded with the COFRAC
(French committee for the accreditation), allows the
research laboratories to prove the quality of their
organization and to assure the quality of their
results.
This report approaches the various stages of
the implementation of a method quality, within a laboratory
realizing tests of electric security on medical
devices.
In a first part, the requirements of the
standard ISO 17025 are clarified within the framework of
this laboratory. In a second part, a methodology of
implementation is proposed by means of a decomposition of
activities under shape of process. Finally, certain elements
are clarified to help in the elaboration of the
textbook(manual worker) quality, procedures, instructions
and associated recordings.
|
|
|
SOMMAIRE
INTRODUCTION
I)
Pré-requis
A)
LES NORMES APPLIQUEES AU TEST DE SECURITE
ELECTRIQUE
B)
L'ACCREDITION
II)
Présentation de la norme 17025
A)
ANALYSE DES EXIGENCES ORGANISATIONNELLES
1.
Organisation de la direction
2.
Elaboration d'un système qualité
3.
Maîtrise de la documentation
4.
Revue des demandes, appel d'offres et contrats
5.
Sous-traitance des essais et des étalonnages
6.
Maîtrise des travaux d'essais ou d'étalonnage non
conformes
7.
Maîtrise des enregistrements
8.
Audits internes
B)
ANALYSE DES EXIGENCES TECHNIQUES
1.
Généralités
2.
Méthodes d'essais et validation
3.
Équipement
4.
Traçabilité du mesurage
5.
Planification des procédures de l'échantillonnage et
manutention des objets
6.
Surveillance de la qualité, des résultats d'essai et
rédaction des rapports
III)
Méthode de mise en place dans un laboratoire
biomédical
A)
PLANIFICATION DU PROJET EN VUE D'UN ACCREDITATION
17025
B)
MACRO PROCESSUS ET CYCLES DE VIE
a)
Réponses au exigences techniques
1.
macro processus global : « évaluation de la
sécurité électrique selon la norme 17025
»
2.
Cycle de vie du test
3.
Le processus matériel de tests
4.
Le processus personnel
b)
Réponses aux exigences organisationnelles
1.
Le processus documentation
2.
Le processus de traitement des non conformités
3.
Le processus des audits internes
C)
METHODOLOGIE DE REDACTION
a)
Le manuel qualité
1.
Première approche : la méthode PIEM
2.
Deuxième approche : la méthode des cycles de
vie
b)
Les procédures
c)
Les instructions
d)
Les enregistrements
IV)
Perspectives
CONCLUSION
BIBLIOGRAPHIE
ANNEXES
INTRODUCTION
L'obligation d'effectuer
des contrôles qualité en milieu hospitalier est apparue.
En effet le décret d'application L 665-5 du 5 décembre
2001 de la Loi du 1 juillet 1998 vient de préciser les
obligations de contrôle qualité des dispositifs
médicaux.
Le défi à
relever pour un service biomédical sera donc, de
démontrer son aptitude à assumer ces exigences
réglementaires légitimes garantissant la
sécurité des patients et des utilisateurs par la mise
en place :
- De méthodes d'essais
et de contrôle qualité rigoureuses
- De processus permettant de
démontrer leur validité (démarche d'assurance
qualité).
Gage d'une
compétence technique et d'une organisation
maîtrisée, l'accréditation, suivant la norme ISO
17025, pourrait être une solution potentielle.
L'accréditation, suivant la définition du Comité
français d'accréditation (COFRAC), vise à
démontrer la compétence d'une entité à
assurer une fonction définie.
La norme ISO 17025,
quant à elle, définit les critères
généraux concernant le fonctionnement d'un laboratoire
d'essais. Celle-ci est basée sur un ensemble de
caractéristiques et d'exigences à la fois
organisationnelle et techniques.
Le test de
sécurité électrique peut tout à fait
rentrer dans ce contexte. Il est définit selon la norme EN
60601, et c'est un contrôle qualité « transversal
» qui s'applique sur de nombreux dispositifs
médicaux.
Ce rapport a donc pour
objectif, d'aider les laboratoires biomédicaux qui voudraient
se lancer dans une démarche d'accréditation. Tout
d'abord, nous définirons le test de sécurité
électrique et la norme EN 60601-1. Dans un deuxième
temps, nous aborderons et expliciterons toutes les exigences de la
norme ISO 17025 (exigences organisationnelles et techniques), nous
définirons ensuite la planification projet à mettre en
œuvre et apporterons des éléments de
réponse concernant l'élaboration du manuel
qualité afin de réaliser un test de
sécurité électrique sur des dispositifs
médicaux selon la norme ISO 17025. Enfin, nous aborderons les
perspectives concernant sa mise en application.
I)
PRE-REQUIS
A)
LES NORMES APPLIQUEES AU TEST DE SECURITE ELECTRIQUE :
La sécurité
dans l'utilisation du courant électrique occupe une place
prépondérante dans les normes. Dans le domaine
médical, pour assurer la sécurité et
éviter tout risque pour le patient et les opérateurs,
les différentes associations normatives ont publié des
prescriptions précises. La norme IEC 601.1 est le document
général complété ou modifié par un
certain nombre de prescriptions spéciales regroupées
dans les Règles Particulières de Sécurité
des Appareils Electromédicaux.
La norme NF EN 60 601-1
détermine les règles générales de la
sécurité électrique des équipements
médicaux. Elle définit les courants de fuite et
courants auxiliaires patient à mesurer. Les valeurs limites de
ces courants sont fixées selon la classification des
appareils. Bon nombre d'entre eux font l'objet de normes
spécifiques qui modifient ou complètent la norme
générale. L'étude combinée de ces normes
permet de développer des procédures de contrôle
de sécurité électrique. Celles-ci regroupent
l'identification de l'équipement, la liste du matériel
nécessaire, le schéma du montage et les
résultats.
Le test de
sécurité électrique est une mesure des
différents courants de fuite et des différents courants
auxiliaires des dispositifs médicaux (annexe I)
B)
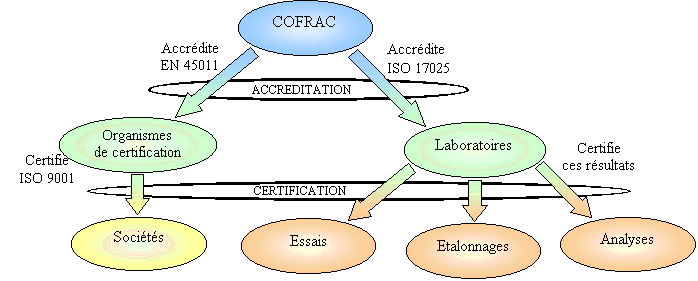
L'ACCREDITION :
Le COFRAC accrédite
entre autres :
- Des organismes de
certification, qui eux même peuvent certifier des organismes
afin de donner la preuve de la qualité de leur
organisation.
- Des laboratoires
d'analyses et d'essais, qui pourront certifier la qualité
de leurs résultats.
II)
PRESENTATION DE LA NORME 17025
La norme ISO 17025 se
divise en deux grandes parties : la première définie
les exigences organisationnelles, la seconde les exigences
techniques.
Cette première approche
de la norme a pour but de la présenter d'une manière
globale afin d'effectuer une synthèse des différentes
exigences. Néanmoins, une étude plus approfondie de la
norme a été réalisée : l'annexe II
reprend chaque paragraphe, en apportant des éléments de
réponses, afin de faciliter sa mise en application, notamment
à l'aide de la méthode PIEM qui définie pour
chaque exigence, les éléments de réponse
à intégrer au manuel qualité .
A)
ANALYSE DES EXIGENCES ORGANISATIONNELLES
Cette première
partie de la norme est divisée en 7 grandes parties
:
1. Organisation du
laboratoire
2. Maîtrise de la
documentation
3. La revue des demandes,
appel d'offres et contrat
4. La gestion de la
sous-traitance des essais et étalonnage
5. La maîtrise des
travaux ou étalonnage non conformes
6. La Maîtrise des
enregistrements
7. Les audits
internes
Les différents
acteurs d'un laboratoire d'essai :
|
Fonction
|
Responsabilité
|
|
La
direction
|
Définie
les objectifs à atteindre
|
|
Un
responsable qualité
|
Doit veiller
à la mise en application et maintien des exigences
organisationnelles
|
|
Un
responsable technique
|
Doit veiller
à la mise en application et maintien des exigences
technique
|
|
Un
responsable métrologie
|
Responsable
de la calibration des appareils de mesure
|
|
Des
opérateurs
|
Réalisation
des essais selon les procédures
|
|
Des
auditeurs
|
Effectuent
les audits internes
|
1.
Organisation de la direction
1.1
Responsabilité juridique (paragraphe 4.1.1)
Ce premier paragraphe
aborde la notion de responsabilité juridique : une
entité juridique est responsable des activités du
laboratoire qui sont décrites dans son manuel
qualité.
1.2
Champs d'application (paragraphe 4.1.2)
Ce paragraphe
définie le contexte dans lequel le laboratoire
évolue :
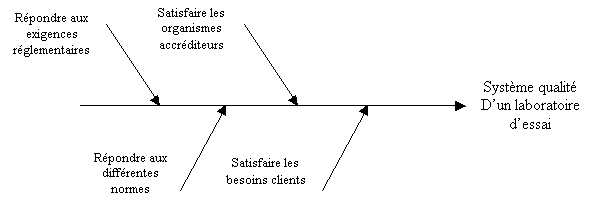
Figure 1 : champs
d'application d'un système qualité
On retrouvera des
réponses à divers endroits du système
qualité.
- L'évaluation du
niveau de satisfaction du laboratoire aux exigences de la
norme est apportée par l'audit interne pour le
management, par les contrôles et les intercomparaisons pour
les exigences technique.
- La conformité
aux besoins clients sera analysée en fonction de
leur demande et de leur contrat.
- Les exigences de
l'organisme d'accréditation doivent faire l'objet,
lorsqu'il en existe, de disposition examinées lors de
contrôles et d'audits technique.
- Les exigences des
autorités réglementaires, elles doivent faire
l'objet d'une veille réglementaire et d'une
vérification périodique permettant d'assurer
qu'elles sont toujours satisfaites.
Si les exigences de la norme,
des clients et de l'organisme de reconnaissance sont clairement
identifiables, il est plus délicat de répondre de
façon exhaustive aux exigences réglementaire qui
peuvent concerner tout à la fois la sécurité sur
le lieu de travail, la détention de certains produits,
...
1.3
Contenu du système de management (paragraphe
4.1.3)
Le laboratoire doit
décrire les activités pour lesquelles le système
qualité s'applique. Cependant, il est courant de rencontrer
des systèmes qualité dont le champ d'application est
plus vaste que le périmètre couvert par la demande
d'accréditation. En effet, même si on accrédite
une partie du laboratoire, il est intéressant de mettre en
œuvre une démarche qualité globale afin de ne pas
exclure le reste du laboratoire.
1.4
Définition des fiches de fonction et des dispositions
concernant l'impartialité, l'intégrité et la
confidentialité (paragraphe 4.14)
Si un organisme d'essais a
d'autres activités, il convient d'identifier les personnes
susceptibles d'être confrontées à un conflit
personnel et de décrire précisément leur
fonction. Cette exigence est applicable uniquement aux laboratoires
faisant partie d'une organisation qui a des activités autres
que les essais ou l'étalonnages.
1.5
Obligations générales du laboratoire (paragraphe
4.1.5)
Cette exigence
définie les éléments nécessaires à
la mise en place d'un système qualité dans un
laboratoire d'essai. Le laboratoire devra donc intégrer dans
son système qualité certains éléments
comme (voir l'annexe II):
- Définir les
responsabilités du personnel d'encadrement ;
- Afficher son
indépendance ;
- Prouver son
impartialité et intégrité ;
- Indiquer son
organisation hiérarchique, fonctionnelle et qualité
;
- Nommer du responsable
qualité ;
- Délimiter la
fonction du responsable qualité ;
Des fiches de
définition de fonction, les dispositions concernant
l'impartialité, l'intégrité et la
confidentialité, les organigrammes, la description des
encadrements organisationnel et techniques mis en place, la
nomination d'un responsable de la fonction qualité et la
désignation des suppléances aideront le laboratoire
à répondre aux exigences.
2.
Elaboration d'un système qualité
2.1
Rédaction d'un système qualité (paragraphe
4.2.1)
Les systèmes
qualité dépendent principalement du type de management
de chaque laboratoire, ils sont à l'image de l'organisation et
des processus du laboratoire. Le degré de détail de la
documentation devra être adapté à sa juste mesure
afin de pouvoir assurer la qualité de la prestation du
laboratoire. Le responsable qualité devra se poser cette
question « quel est le bon degré
de détail de la documentation ? ».
2.2
Rapport entre le manuel qualité et la politique qualité
(paragraphe 4.2.2)
La politique qualité
est le point le plus important d'un système qualité.
Souvent définie sous comme un simple engagement, une politique
qualité doit déterminer les objectifs
généraux que le laboratoire doit atteindre qui servent
eux-mêmes à fixer les objectifs qualité, dont les
trois propriétés fondamentales sont les suivantes :
être réaliste, mesurables et limités dans le
temps.
La rédaction de la
déclaration de la politique qualité est une
étape importante et ce notamment d'un point de vue
psychologique. Cette déclaration doit être un compromis
d'ambition et de réalisme. Elle devra être claire,
simple et accessible à l'ensemble du personnel et
répondre aux exigences normatives sans être
dénaturée. La politique qualité d'un laboratoire
sera définie selon la roue de « Deming » ou PDCA
décrit dans la figure 2.
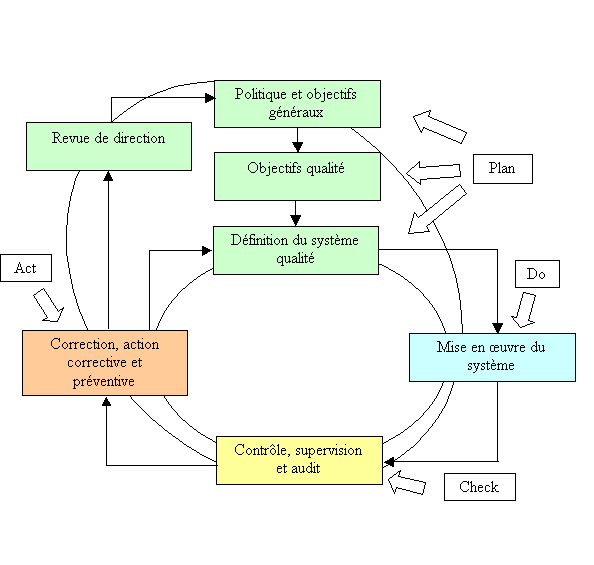
Figure 2 : fonctionnement
d'un système qualité [9]
2.3
Rapport entre le manuel qualité et les procédures de
soutien (paragraphe 4.2.3)
Le manuel
qualité est « la carte routière du système
», il est donc important de bien choisir son architecture
(« pyramidale » ou « en râteau
»).
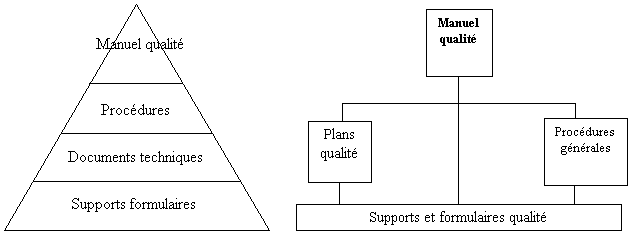
Figure 3 : architecture
d'un système qualité[9]
Remarque : Pour
l'élaboration de notre manuel qualité qui sera
développé dans la troisième partie, nous avons
choisi l'architecture pyramidale
2.4
Délimitation des rôles (paragraphe 4.2.4)
Ce paragraphe
définit les missions des différents responsables. Pour
simplifier, on peut dire que le responsable qualité a la
charge de piloter toutes les actions relatives au chapitre 4 de la
norme (Prescriptions relatives au management) et le directeur
technique celle des actions relatives au chapitre 5 (Prescriptions
techniques). Ce chapitre impose au laboratoire de décrire ses
missions en démontrant la séparation harmonieuse
adoptée pour concilier cette union difficile entre
qualité et technique.
3.
Maîtrise de la documentation
3.1
Généralités (Paragraphe 4.3)
Le laboratoire doit
disposer d'un système documentaire et de modalités de
gestion documentaire explicites. Pour simplifier et clarifier la
formalisation des documents, on rédigera une procédure
pour la « maîtrise de la documentation interne », la
procédure traitant des documents externes est en
général suffisamment simple pour en faire un court
paragraphe dans le manuel qualité.
3.2
Approbation et diffusion des documents (paragraphe
4.3.2)
Ces 3 paragraphes abordent
la gestion documentaire du système qualité, elle
comprend :
- Revoir et approuver les
documents ;
- Définir la liste
des personnels autorisés à revoir et approuver les
documents ;
- Tenir à jour la
liste des document en vigueur ;
- Gérer la liste
de diffusion de documents ;
- Définir les
modalités de diffusion ;
- Mettre en place une
revue périodique de la documentation ;
- Retirer des documents
périmés ;
- Marquer des documents
à conservés à des fin légales ou de
traçabilité ;
- Identifier tous les
documents ;
Remarque : Pour
répondre aux différentes modalités de
rédaction des procédures choisies, il est possible
d'intégrer la totalité des exigences
méthodologiques du paragraphe au sein d'un processus, et de le
définir en sous processus traitants de thème comme la
diffusion et le retrait des périmés d'une part, la
codification de la documentation, d'autre part.
3.3
Modification des documents (paragraphe 4.3.3)
Cette exigence aborde la
modification de la documentation, elle :
- Définie la revue
et l'approbation des modifications des documents ;
- Permet d'accéder
aux informations pour la validation de ces modifications
;
- Identifie les
modifications ;
- Définit les
modalité afin de modifier manuellement la documentation
;
- Aborde les
modalités pour réaliser les modifications manuelles
;
- Explicite la
rémission d'un document après modification manuelle
;
- Décrit les
modifications de la documentation électronique.
Si une modification est
apportée à un document, l'utilisateur doit pouvoir
identifier le plus rapidement possible quels éléments
ont été modifiés par rapport aux pratiques
précédentes.
4.
Revue des demandes, appel d'offres et contrats
4.1
Approche revue de contrat (paragraphe 4.4.1)
L'exigence « revue de
contrat » est une formalisation des accords entre les
laboratoires qui a été conçue pour éviter
qu'ils effectuent des essais sans en connaître l'étendue
exacte. Son objectif principal est d'apporter la réponse
adéquate aux demandes des clients tout en s'assurant que le
laboratoire dispose bien des ressources nécessaires pour le
satisfaire, que ce soit au niveau de la disponibilité des
équipements, des consommables, des locaux, qu'à celui
de la compétence de son personnel et cela dès que le
client intègre les exigences de délai.
Pour toutes relations entre
un client et un fournisseur, la décision commune sur le
contenu de la prestation à réaliser doit faire l'objet
d'un accord, en particulier lors du remaniement de la demande
initiale et des discussions entre les deux parties.
La mise en place d'un
planning d'analyses permettant d'évaluer la charge des quatre
composantes principales des moyens du laboratoire : le
matériel, les locaux et les consommables.
L'enregistrement, l'estimation
des demandes et l'évaluation de la capacité du
laboratoire à répondre aux exigences clients doivent
précéder celle de la satisfaction finale.
4.2
Conservation de l'enregistrement des revues (paragraphe
4.4.2)
La revue permettant
d'attester que le laboratoire a bien évolué sa
capacité à répondre aux exigences, aux besoins
et attentes du client doit être enregistrée et
conservée, ainsi que l'ensemble des relations orales ou
écrites avec le client concernant sa prestation.
4.3
Attribut de la revue (paragraphe 4.4.3)
Le laboratoire ne doit
accepter aucun contrat avec une société sous-traitante
s'il n'est pas sûr qu'elle puisse répondre correctement
aux demandes, que ce soit en terme de délai, de
compétence, etc.
4.4
Obligations avec le client (paragraphe 4.4.4)
Le laboratoire doit tenir
informé le client des écarts qui conduisent à
des modifications de contrat. Il faudra donc expliciter d'une
manière ou d'une autre les moyens choisis par le laboratoire
pour informer le client d'écart par rapport au contrat. Les
responsabilité correspondantes devront être
décrites.
4.5
Répétition du processus de revue de contrat (paragraphe
4.4.5)
Un processus de revue
permettra aux deux parties de se mettre formellement d'accord sur la
réalisation du contrat. On pourra éventuellement y
associer un avenant.
5.
Sous-traitance des essais et des
étalonnages
5.1
Généralités (paragraphe 4.5.1)
La sous-traitance est une
opération contractuelle pour laquelle un laboratoire confie la
totalité ou une partie des travaux à un autre
laboratoire au profit direct du client. Des critères de
compétences devront être fixés par le contractant
en fonction de la nature et de l'étendue de la sous-traitance,
ceux-ci devant faire l'objet d'une vérification
appropriée.
Si le laboratoire n'a pas les
compétences nécessaires pour exécuter une
demande et qu'il doit sous-traiter la totalité des essais, de
manière continue ou momentanée, les critères
choisis devront être ceux de la norme. Dans tous les cas, le
résultat de la sous-traitance devra être
contrôlable.
5.2
Sous-Traitance (paragraphe 4.5.2)
Le laboratoire qui
décide de sous-traiter doit prévenir le client et
obtenir son accord au risque d'effectuer l'essai dans un second
laboratoire choisi par le client.
5.3
Responsabilité des travaux (paragraphe 4.5.3)
Lorsqu'une prestation
d'essais est soumise à un laboratoire, il paraît logique
que celui-ci prenne en charge la responsabilité du contrat,
sauf s'il lui a été imposé. Le laboratoire doit
expliciter, son attitude et les dispositions éventuelles qu'il
applique quand le sous-traitant lui est imposé.
5.4
Enregistrement des activités confiées aux
sous-traitants (paragraphe 4.5.4)
Le sous-traitant
accrédité apportera la preuve de sa conformité
non seulement par la présentation de son attestation
d'accréditation, mais également par celle de son annexe
technique qui assure la compétence au titre des travaux sous
traités.
5.5
Achats de services et relation avec le client
5.5.1
Procédure d'achat (paragraphe 4.6.1)
L'exigence concerne l'achat
de service et de fournitures, la réception et le stockage de
ces fournitures.
5.5.2
Produits et services achetés ( paragraphe
4.6.2)
Le laboratoire doit
définir le niveau de maîtrise qu'il entend mettre en
place sur les fourniture, les produits consommable achetés. Le
niveau de contrôle dépend du risque que l'on peut
prendre et donc de la criticité de la conformité du
produit dans son utilisation.
5.5.3
Contenu des documents d'achats (paragraphe 4.6.3)
On peut définir les
document d'achat comme un ensemble de documents communiqués
aux fournisseurs pour les aider à effectuer leur propre revue
de contrat. Dans la mesure où le
bon d'achat constitue le
contrat entre le laboratoire et son fournisseur, il doit contenir
l'ensemble des spécification formalisant les exigences du
laboratoire en matière de qualité. Les modalités
de revue, c'est à dire de relecture critique et d'approbation
des bons de commande, doivent être définies ,
formalisées, mise en œuvre et faire l'objet d'une trace
écrite.
5.5.4
Evaluation des fournisseurs de produits consommables, fournitures et
services critiques (paragraphe 4.6.4)
L'exigence demande au
laboratoire d'évaluer ses fournisseurs de produits
consommables, fournitures et services critiques qui pourraient
affecter la qualité des essais. Ces évaluation devront
faire l'objet d'enregistrements.
5.6
Services à la clientèle (paragraphe 4.7)
Le client est en position
privilégiée. Afin d'évaluer la satisfaction
client le laboratoire doit mettre en place des évaluations
afin de mesurer la satisfaction des clients.
5.7
Réclamations (paragraphe 4.8)
Les réclamations
peuvent provenir de clients mais également d'autres parties.
Le processus mis en place doit intégrer ces contraintes.
Ensuite, une procédure de réclamation devrait
débuter par deux tests. Le premier consiste à
vérifier que la réclamation est justifiée, le
second permet d'évaluer s'il ne s'agit pas d'une forme de
pression tentant de mettre en péril l'impartialité du
laboratoire.
Lorsque ces deux tests ont
été effectués, le laboratoire se retrouve devant
un cas de traitement de non conformité. Il est important de
relier ensuite en retour le traitement de la non-conformité au
processus de traitement de la réclamation qui doit
prévoir une information au client des dispositions
prises.
6.
Maîtrise des travaux d'essais ou d'étalonnage non
conformes
6.1
Politique de traitement des non-conformités (paragraphe
4.9.1)
Ce point est une clef de
l'évolution du système qualité. La norme impose
à tous les laboratoire d'évaluer l'importance de la non
conformité. Il s'agit d'équilibrer soigneusement le
traitement effectué, en veillant à l'harmonisation du
prix et de la qualité de traitement de la
non-conformité.
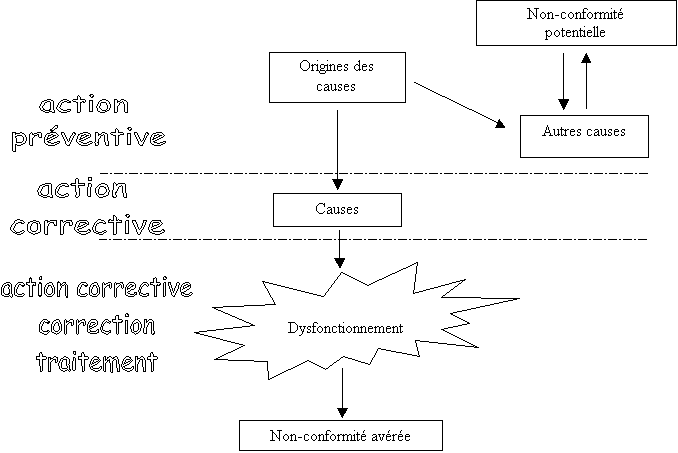
Figure 4 :
Non-conformité, action corrective et action
préventive[9]
6.2
Attribution des responsabilités en matière de
traitement des non-conformités (paragraphe
4.9.2)
La procédure de
non-conformité doit s'achever par un test évaluant la
faculté de l'écart à se reproduire. Une
réponse positive à ce test doit conduire au lancement
de la procédure d'action corrective décrite dans les
paragraphes suivants. Si le test est négatif, il suffit
d'enregistrer cette conclusion.
6.3
Actions correctives
6.3.1
Identification (paragraphe 4.10.1)
Les problèmes
identifiés peuvent être des écarts par rapport au
système qualité ou des écarts techniques. Il
s'ensuivra une prise de décision sur la validité de
l'action choisie par la personne la plus compétente, par,
exemple, le responsable qualité dans un cas, le responsable
technique dans l'autre. Comme pour traiter les
non-conformités, une procédure s'avère
indispensable en tant que document. Il convient de décrire les
responsabilités désignées pour l'ensemble des
items présentés dans les paragraphes qui suivent ;
analyse des causes, choix des actions, la mise en œuvre de ces
actions et la surveillance de leur efficacité.
6.3.2
Analyse des causes (paragraphe 4.10.2)
Cette enquête
est une étape importante du processus. Pour la
réaliser, il existe des outils comme, le diagramme
causes-effets. Selon cette méthode, tout effet est dû
à une cause ou à une combinaison de causes que l'on
peut classer dans sept catégories. Elle permet d'avoir une
approche très structuré d'analyse des
causes.
Cependant l'observation
physique du problème ne pourra être remplacée et
reste donc complètement indispensable. En revanche, elle
évite de ne prendre en compte que la partie immergé de
l'iceberg. Si l'utilisation de ces outils est systématique, il
est souhaitable que le laboratoire en décrive les principes.
L'enregistrement des résultats de cette analyse devra
être formalisée de telle sorte que, si la solution
choisie par la suite se révèle n'être pas ou pas
suffisamment efficace, le travail effectué à cette
phase n'ait plus qu'à être revalidé plutôt
que repris en totalité.
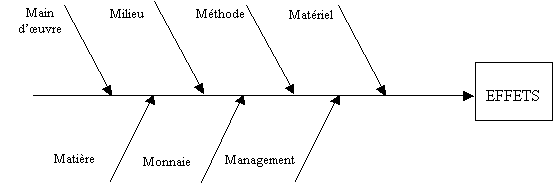
Figure 5 : diagramme des
7 M ou « cause effet » [9]
6.3.3 Choix et mise en œuvre d'actions correctives (paragraphe
4.10.3)
La méthodologie
à suivre est totalement imposée par la norme :
décision de lancer une action corrective et analyse des causes
conduit à identifier toutes les actions correctives
correspondantes puis à choisir la plus pertinente. Il convient
alors de formaliser les modifications de procédures induites
par ces actions correctives. On pourra aussi, si l'on a du recul,
mettre en œuvre une hiérarchisation des causes en
s'appuyant sur le principe selon lequel il est inutile de travailler
sur une cause dont l'occurrence est particulièrement faible ou
dont la contribution à l'effet global constaté est
faible.
Le gestion des
non-conformités rentre de nouveau dans le cadre d'un plan
d'amélioration continue de la qualité :
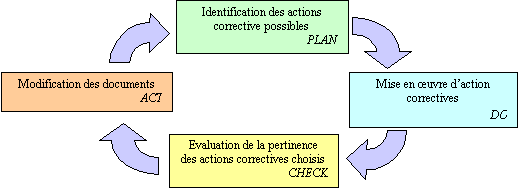
Figure 6 : gestion des
actions correctives
6.3.4 Surveillance des actions correctives (paragraphe
4.10.4)
La mise en place d'un
comité de suivi permettra :
- d'analyser les
non-conformités qui se sont produites ;
- de valider le choix
d'action corrective engagée ;
- de fixer un
délai pour le suivi des actions correctives en fonction du
degré d'occurrence de la cause et de l'effet ;
- de gérer les
détails des actions correctives mise en place en validant
leur efficacité c'est à dire en vérifiant que
l'effet généré ne s'est pas reproduit pendant
le délai choisi.
6.3.5 Audits complémentaires (paragraphe
4.10.5)
Ces audits portent
spécifiquement sur les non-conformités «
stratégiques ».
6.3.6 Actions préventives (paragraphe 4.11.1)
Les plans d'actions
auxquels la norme fait allusion, sont des documents contenant des
actions pour lesquelles des responsabilité et des
délais ont été définis. On peut y
associer les moyens et les éléments permettant la
vérification de la mise en œuvre des actions.
L'identification des actions préventives pourra être
confiée au comité de suivi . L'analyse de toutes les
non-conformités et actions mise en œuvre apportera des
idées pertinentes.
6.3.7 Procédures relatives aux actions préventives
(paragraphe 4.11.2)
Cette exigence a pour but
d'apporter tous les éléments nécessaires
à l'organisation et à la mise en place de ces
procédures.
Remarque : la procédure
d'action corrective peut être couplée à celle
requise par ce paragraphe et les méthodologies des deux
procédures peuvent être semblables. Il suffit pour cela
que la donnée d'entrée de la procédure soit de
type « non-conformité réelle ou potentielle
».
7.
Maîtrise des enregistrements
7.1
Définition des enregistrements (paragraphe
4.12.1)
Ce paragraphe
définit les modalités liées à la gestion
de la documentation :
- La gestion des
enregistrements ;
- La lisibilité,
le stockage et la conservation des enregistrements ;
- La durée de
conservation des enregistrements ;
- La sûreté
et la confidentialité de la conservation ;
- La protection et
sauvegarde des enregistrement informatiques.
Un formulaire d'enregistrement
est un support de données, des enregistrements et des
résultats. Ces dispositions s'appliquent également aux
formulaires correspondant à chacun des documents : manuel,
procédure, instruction ou mode opératoire. Leur nombre
et leur nature sont peu soumis à variation, si ce n'est pour
prendre en compte telle ou telle évolution du document. En
revanche, les enregistrements sont en quantité
évolutive car à chaque travail du laboratoire
correspond une retranscription.
Par conséquent tout
processus « enregistrements » devra contenir les
informations suivantes :
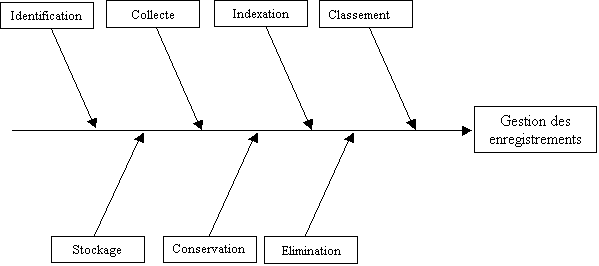
Figure 7 : diagramme
"cause effet" des enregistrements
7.2
Enregistrements techniques (paragraphe 4.12.2)
L'objectif principal des
enregistrements « en assurance qualité » est de
permettre la traçabilité et de retrouver l'historique
des informations.
8.
Audits internes
8.1
Fréquence des audits internes (paragraphe
4.13.1)
L'objectif de ce processus
est de réaliser non pas un audit sur l'ensemble des
activités du laboratoire comme le fait déjà
l'organisme accréditeur mais de faire de nombreux audits
beaucoup plus courts, dont l'ensemble couvre la totalité du
système et des activités techniques. Pour cela le
laboratoire devra définir un programme d'audits. Un
système d'audit se compose :
- d'une procédure
d'audit, de la qualification des audits internes
- de l'élaboration
structurée et documentée d'un programme
d'audit.
8.2
Enregistrement des résultats de l'audit (paragraphe
4.13.2)
Tout écart
constaté doit induire à la recherche des causes qui en
sont à l'origine, des conséquences qu'il engendre et
à informer le client si les résultats qui lui ont
été communiqués sont entachés d'un doute.
Il est recommandé d'effectuer un audit par la «
traçabilité » qui consiste, en partant soit d'une
demande client (traçabilité aval), soit d'un rapport
d'essai (traçabilité amont) , à analyser
l'ensemble des processus, la documentation qui leur est
associée et leur mise en œuvre.
Si le processus d'action
est bien construit, il n'est pas nécessaire de
répéter cette exigence. Il suffit de connecter la
procédure d'audit interne à la procédure
d'action corrective dès que l'on constate un écart de
management du laboratoire ou dans les prescriptions techniques. Le
seul élément complémentaire est l'information au
client que le manuel qualité peut traiter.
8.3
Rapport d'audit (paragraphe 4.13.3)
La trace de l'audit
doit être assurée. Le rapport devra contenir l'ensemble
de ses données ou y faire référence. Si l'on
choisit d'enregistrer les écarts constatés sur les
fiches de non-conformité et de les gérer comme à
l'accoutumée, le rapport d'audit peut se contenter de
reprendre « les identifiants » de l'audit (voir annexe
II)
8.4
Efficacité des actions correctives (paragraphe
4.13.4)
Suite à ces
audits, le laboratoire doit prendre en compte les
non-conformités et s'assurer de leur efficacité.
Néanmoins, le suivi de l'efficacité des actions
correctives suffit pour remplir ce rôle.
8.5
Contenu de la revue de direction (paragraphe 4.14.1)
Le but d'une revue de
direction est d'afficher une politique, de la traduire en objectifs
précis et de dresser ensuite un bilan des résultats. Au
cour de cette revue, il s'agit de présenter les
résultats de la période précédente en y
intégrant l'ensemble des éléments exigés
par la norme pour en faire une analyse en réunion de direction
afin d'évaluer l'efficacité des dispositions prises par
le système qualité. L'objectif de ce processus est de
prendre des décisions en fonctions des résultats de
cette analyse et de dresser un plan d'actions pour la période
suivante et une éventuelle révision de la politique
qualité et des objectifs correspondants.
8.6
Contenu des revues de direction (paragraphe 4.14.2)
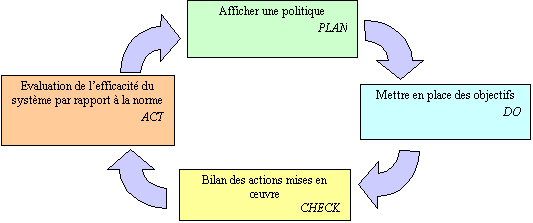
Figure 8 : principe
d'une revue de direction
Si suite à une revue
de direction, des décisions ont été prises et
qu'un plan d'action a été lancé, il faut alors
s'assurer et rendre compte à la direction de la mise en
œuvre des actions afin de vérifier qu'elles produisent
l'effet escompté dans les délais choisis.
B)
ANALYSE DES EXIGENCES TECHNIQUES
Les exigences techniques
abordent l'ensemble des processus se rapportant au déroulement
des essais, elles sont divisées en 6 parties :
1.
Généralités
2. méthodes d'essais et
validation
3. les
équipements
4. traçabilité
du mesurage
5. planification des
procédures et manutention des objets
6. surveillance de la
qualité des résultats d'essai et rédaction des
rapports de résultats.
Ces exigences techniques
abordent 3 types de tests :
- La réalisation
d'essai sur des produits ;
- L'étalonnage de
dispositifs ;
- L'échantillonnage
sur des grandes quantités de produit.
Pour le test de
sécurité électrique, seul les exigences
dédiées à la réalisation d'essai devront
être prises en compte, ce qui nous permettra par exemple d'en
éviter certaines (ex : paragraphe 5.7). Dans cette analyse,
les spécificités du test de sécurité
électrique sont abordées.
Le matériel
nécessaire à la réalisation d'un test de
sécurité électrique :
|
DESIGNATION
|
CATEGORIE(défini
paragraphe 5.5.4)
|
FONCTION
|
|
Testeur de
sécurité électrique répondant
à la norme EN 60601
|
Matériel
d'essai
|
Réalise
entièrement l'essai selon la norme
|
|
Tapis
antistatique
|
Matériel
intermédiaire
|
Isole le
dispositif à tester
|
|
Thermomètre
|
Matériel
de mesure
|
Mesure des
conditions d'environnement
|
|
Hygromètre
|
Matériel
de mesure
|
Mesure des
conditions d'environnement
|
|
Baromètre
|
Matériel
de mesure
|
Mesure des
conditions d'environnement
|
|
Accessoires
|
Matériel
intermédiaire
|
Relie les
parties appliquées des différents dispositifs
médicaux
|
1. Généralités
1.1
Analyse et mesure de l'incertitude (paragraphe 5.1.1)
Ce paragraphe
définit les nombreux facteurs qui déterminent
l'exactitude et la fiabilité des essais effectués par
un laboratoire. Ces facteurs peuvent comprendre des
éléments provenant
- de facteurs humains
(para 5.2);
- des installations et
conditions ambiantes (para 5.3);
- des méthodes
d'essai et d'étalonnage et de la validation des
méthodes (para 5.4);
- de l'équipement
(para 5.5);
- de la
traçabilité du mesurage (para 5.6);
- de
l'échantillonnage (para 5.7);
- de la manutention des
objets d'essai et d'étalonnage (para 5.8).
Ce paragraphe introduit les
prescriptions et indique les paragraphe 5.2 à 5.8 dans
lesquels sont traités les facteurs techniques pouvant influer
sur la qualité des essais.. il est complété par
des prescriptions sur la maîtrise et la surveillance de la
qualité, d'une part, et sur le contenu du rapport d'essai,
d'autre part.
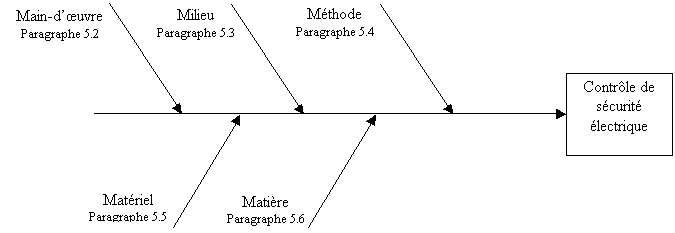
Figure 9 : diagramme 5 M
sur le contrôle de la sécurité
électrique
Pour répondre
à cette exigence dans le manuel qualité, il est
possible de dresser une liste de tous les éléments
susceptibles d'influencer la qualité des essais.
1.2
Divers facteurs à prendre en compte (paragraphe
5.1.2)
Cette exigence traite en
premier lieu de la maîtrise des incertitudes. Il s'agit donc
d'adapter les performances au domaine technique d'essai et
d'acquérir un niveau d'exigence.
Les facteurs qui rentrent
en compte dans le test de sécurité électrique
des dispositifs médicaux sont :
- l'opérateur
- le matériel
permettant d'effectuer l'essai (testeur + accessoires)
- l'environnement
- le dispositif
médical à tester
Ces facteurs vont
être développés dans les exigences
ci-dessous.
1.3
Personnel
1.3.1 Compétence du personnel technique (paragraphe
5.2.1)
Cette exigence a pour but
de définir les différents types d'activités du
personnel :
- La réalisation
des essais ;
- Evaluation des
résultats ;
- Validation des
rapports.
De plus, elle impose la
supervision du personnel. Par, exemple, un planning de formation peut
être prévu. A partir de l'évaluation de niveau de
départ de la personne, il permettra d'étaler les
formations dans le temps pour amener la personne au niveau de
compétence souhaité.
Les compétences des
divers opérateurs devront être contenus dans un dossier.
Néanmoins, la norme n'impose pas de niveau de formation, ce
qui implique, que le laboratoire doit décider des
spécifications concernant chaque type de personnel du
laboratoire.
1.3.2 Description des objectifs de la formation du personnel
(paragraphe 5.2.2)
Les engagements de la
direction en matière de formation et la stratégie
adoptée doivent être déployés
concrètement dans le programme de formation mis en place. Ces
engagements seront traduits dans l'identification des besoins en
formation et dans la mise en œuvre de cette formation. Toutes
les traces de ces actions seront conservées pour prouver leur
mise en place.
1.3.3 Choix des collaborateurs (paragraphe 5.2.3)
De la même
façon que pour les collaborateurs en cours de formation, la
supervision des personnes employées à titre temporaire
est requise. Il paraît donc difficile de se priver d'une
procédure permettant au personnel d'appoint d'avoir
l'information minimale nécessaire sur les aspects techniques
et de management du système qualité à mettre en
œuvre. La création d'une forme de livret d'accueil
facilitera la diffusion des documents pertinents. Néanmoins,
ces personnes devront être accompagnées dans leurs
premiers pas techniques jusqu'à l'obtention de l'assurance de
leur compétence, tout en conservant à la fois la preuve
des supervisions réalisées.
1.3.4 Description des fonctions du personnel (paragraphe
5.2.4)
Une fiche devra
définir précisément les responsabilités
des différentes personnes au sein du laboratoire. On
s'attachera à définir les aspects liés au flux
technique et les aspects liées au flux
administratif.
1.3.5 Habilitation à délivrer des rapports
d'essai (paragraphe 5.2.5)
Ce paragraphe traite de la
conservation des éléments permettant d'assurer la
qualification du personnel, d'une part et des enregistrements des
autorisations délivrées par la direction qui s'engage
sur cette compétence d'autre part. Ces éléments
pourront être rédigées dans une « fiche
d'habilitation » qui précisera par exemple les essais
pour lesquelles un technicien a été reconnu
compétent. Elle devra être co-signée par la
direction et par le titulaire attestant d'une part l'engagement de la
direction au regard du contenu de la fiche, et d'autre part, la prise
de connaissance des informations par le titulaire.
1.4
Installations et conditions ambiantes
1.4.1 Installations d'essais et conditions ambiantes (paragraphe
5.3.1)
Cette exigence concerne non
exclusivement les sources d'énergie, l'éclairage et les
conditions ambiantes.
Le paragraphe 10 de la norme
EN 60-601 définit ses conditions d'environnement :
- Température
ambiante doit être comprise entre 10 et
40°C
- Humidité
relative doit être comprise entre 30 et 70%
- La pression
atmosphérique doit être comprise entre 700 hPa et
1060 hPa.
- L'alimentation
électrique devra être maîtrisée (mesure
des fluctuations du réseau)
1.4.2 Surveillance des conditions ambiantes (paragraphe
5.3.2)
Après avoir
défini les caractéristiques d'environnement pouvant
influencer la qualité des essais, le laboratoire doit
:
- Prévenir la
« variation » par la maîtrise ;
- Contrôler
« la valeur » par la surveillance ;
- Prouver leur
maîtrise et leur surveillance par
l'enregistrement.
Le laboratoire doit
décider de l'arrêt lorsque les conditions ambiantes
dépassent les niveaux acceptables. Il est donc important de
déterminer l'autorité qui pourra prendre une telle
décision. Cette exigence suppose que la mesure des conditions
ambiantes soit réalisée par des instruments
étalonnés.
1.4.3 Séparation des éléments (paragraphe
5.3.3)
Il s'agit ici de trouver le
meilleur compromis entre l'exigence d'ergonomie qui implique de
rapprocher certaines zones d'essais et celle de manutention d'objet
en séparant celles qui présentent des caractères
incompatibles.
Dans notre cas on s'attachera
surtout à vérifier la compatibilité
électromagnétique, ainsi que les courants de fuites
hautes fréquences lors du test de sécurité
électrique des bistouris.
1.4.4 Réglementation de l'accès aux zones d'essais
(paragraphe 5.3.4)
Le laboratoire mettra en
place les moyens les plus adaptées en ce qui concerne
l'accès aux bancs de test.
Pour la sécurité
électrique, un simple panneau limitant l'accès aux
personnes du laboratoire et aux visiteurs accompagnés pourrait
suffire.
1.4.5 Entretien du laboratoire (paragraphe 5.3.5)
Le domaine
d'activité et les risques générés par un
mauvais entretien doivent être pris en compte pour choisir les
procédures d'entretien appropriées.
Si on utilise la
sous-traitance il sera important de définir les
responsabilités de nettoyage entre le personnel du laboratoire
et le personnel extérieur, d'une part, l'engagement de
confidentialité que le laboratoire doit obtenir de cette
société de sous-traitance d'autre part.
2.
Méthodes d'essais et validation
2.1
Application des méthodes d'essais (paragraphe
5.4.1)
Cette exigence aborde ici
deux points qui seront développés ultérieurement
: l'estimation de l'incertitude et les techniques statistiques.
Quatre types de documents sont donc exigés :
|
Type
d'action
|
Type
de document
|
|
Les
méthodes d'essais
|
document
opérationnel et de réalisation des
essais
|
|
Les
procédures d'essais
|
documents
organisationnels explicitant le partage des
responsabilités entre les différents secteurs
du laboratoire
|
|
Les
instructions d'utilisation du
matériel
|
document
organisationnels de type notice interne
|
|
Les
instructions de manutention et de préparation des
échantillons
|
documents
opérationnels de manipulation des objets soumis
à essais
|
L'ensemble de ces documents
opérationnels doit bien sûr être
géré et mis à disposition du
personnel.
Cette exigence fait
ressortir trois types de mode opératoire :
- Les modes
opératoires d'essai ;
- Les mode
opératoires de manutention et de préparation des
objets
- Les modes
opératoires d'utilisation des objets.
2.2
Sélection des méthodes d'essai (paragraphe
5.4.2)
Cette exigence implique que
les méthodes d'essai permettent de :
- De répondre au
besoins du client ;
- D'intégrer les
normes et textes réglementaires ;
- De sélectionner
des méthodes non imposées par le client
;
- D'utiliser des
méthodes développés ou adoptées par le
laboratoire ;
- D'informer le client
sur la méthode utilisée ;
- De confirmer de
l'aptitude du laboratoire ;
- D'informer le
client.
2.3
Méthodes développées par le
laboratoire(paragraphe 5.4.3)
Ce paragraphe
définit la :
- Planification du
développement de méthodes
- Qualification du
personnel chargé du développement de
méthodes
- Mise à jour des
plans
- Communication des plans
de développement mis à jour
Cette exigence s'applique aux
laboratoires qui réalisent par exemple des essais de
performance de produits. Elle oblige les laboratoires qui font de la
recherche et développement à élaborer un plan
afin de revalidée des méthodes constamment remises
à jour.
2.4
Méthodes non normalisées (paragraphe
5.4.4)
Si le laboratoire effectue
des essais non définis par une norme, le laboratoire doit
élaborer et valider ses méthodes, informer le client
avant mettre en œuvre sa méthodologie.
Notre laboratoire ne sera
pas concerné par cette exigence, la norme EN 60601-1
définie très clairement les exigences concernant la
sécurité électrique des dispositifs
médicaux.
2.5
Validation des méthodes (paragraphe 5.4.5)
Avant de se lancer
dans le processus de validation d'une méthode interne, il
convient d'en évaluer l'opportunité technique et
financière. Les méthodologies ne peuvent donc
être appliquées qu'une fois leur opportunité
confirmée, qui relève d'une décision politique
de la direction. La laboratoire pourra utiliser une procédure
simplifiée lorsque les modifications par rapport au texte de
référence sont mineures, c'est à dire lorsqu'il
s'agit d'une simple adaptation.
Pour notre
laboratoire, les méthodes employées sont celles
décrites par la norme EN 60601-1, qui définie les tests
à réaliser ainsi que les valeurs limites. Le
laboratoire ne devra donc pas élaborer de méthodes
internes, ces exigences ne sont pas à prendre en compte pour
la mise en œuvre de ces essais de sécurité
électrique.
2.6
Estimation de l'incertitude de mesure (paragraphe
5.4.6)
De même que
pour la validation des méthodes, l'estimation de l'incertitude
est un travail difficile nécessitant la maîtrise d'un
certains nombre d'éléments théoriques et
statistiques. Les normes XP X 07-020 et XP X 07-021 seront d'un grand
secours.
En ce qui concerne la
sécurité électrique, les fournisseurs pourra
fournir l'incertitude de mesure des ces appareils de
tests.
2.7
Maîtrise des données (paragraphe 5.4.7)
La
vérification des calculs et transfert de données peut
être effectuée à différents niveaux
d'organisation. Elle peut être réalisée en
autocontrôle par le technicien ou par un responsable technique
qui analyserait données initiales et données finales.
Les logiciels, ordinateurs et appareils automatisées, doivent
être traités, du point de vue de la documentation
associée, de la même façon que d'autres
équipements : notice d'utilisation, entretien, conditions
ambiantes d'utilisation ...
3.
Équipement
3.1
Pour un bon usage de l'équipement (paragraphe
5.5.1)
Un inventaire des
équipements, des essais auxquels ils sont destinés et
des prescriptions correspondantes de mesurage, permettra d'assurer
que le laboratoire dispose bien de l'ensemble de la
traçabilité nécessaires à
l'étendue de l'accréditation qu'il demande.
Le laboratoire doit
également choisir les modalités d'étalonnage, de
vérification, de calibrage ou de contrôle
appropriés pour assurer que l'équipement est
suffisamment maîtrisé.
3.2
Vérification de l'équipement (paragraphe
5.5.2)
La
périodicité des programmes d'étalonnages, c'est
à dire les planning, devrait être choisie par le
laboratoire en fonction des dérives connues ou
supposées des équipements utilisés.
La norme impose une
vérification avant mise en service, par conséquent,
cette exigence s'appliquera aussi après une réparation
...
Pour les testeurs de
sécurité électrique, la
périodicité est de 1 an, celle-ci pourra être
éventuellement raccourcie en fonction de l'utilisation de
l'équipement.
Le
référentiel porte aussi sur le « contrôle en
service », les différents autotests
réalisées lors de la mise en service du testeur
répondront à cette exigence.
3.3
Liens entre le personnel et l'usage de l'équipement
(paragraphe 5.5.3)
Cette exigence montre
que la maîtrise de la qualité du laboratoire va
au-delà de la simple juxtaposition des principes suivants :
« équipement, personnel, locaux et consommables »,
car il existe de nombreux liens entre tous ces
éléments. Cette exigence porte plus
particulièrement sur la relation entre la matériel et
le personnel autorisé à l'utiliser.
Le laboratoire doit
donc mettre en place des modes opératoires intitulés
« utilisation et entretien de ... » disposant d'un sommaire
standardisé. Lors de la réalisation d'instructions
internes d'utilisation des équipements, il est indispensable
d'identifier les raisons qui justifient leur rédaction. Le
meilleur moyen est de faire rédiger ces modes
opératoires d'utilisation par le technicien qui
relèvera les successions des actions qu'il conduit au fur et
à mesure.
3.4
Identification des équipements (paragraphe
5.5.4)
Pour mettre en
œuvre cette exigence, on utilisera un numéro
d'identification interne plutôt que le numéro de
série fournit par le fabricant afin d'éviter
éventuellement des doublons.
On distingue trois types
d'équipement qui peuvent être définis de la
manière suivante :
|
Le
matériel de mesure
|
matériel
fournissant ou représentant une valeur raccordable
directement ou non aux étalons nationaux ou à
des matériaux de référence
certifiés.
|
|
Le
matériel d'analyse ou d'essai
|
appareils qui
devront être calibrés et contrôlés
avec les moyens les plus appropriés
représentant l'état de l'art
|
|
Le
matériel intermédiaire
|
matériel
ne fournissant pas de résultat ou ne
représentant pas une mesure
matérialisée, mais dont la qualité
participe à la qualité du résultat
final
|
3.5
Identification des équipements (paragraphe
5.5.5)
Cette exigence
définie ce qu'on appelle dans la plupart des cas une fiche de
vie. Ce paragraphe regroupe les éléments
d'identité de l'équipement, les éléments
de suivi de la conformité de l'équipement, les
éléments de maintenance visant à maintenir la
conformités de l'équipement aux spécifications
initiales. Il serai pertinent de mettre en œuvre un suivi des
différents coûts associés à cette
équipement.
Pour répondre à
la norme on pourra donc mettre en place 4 fiches :
- La fiche
signalétique de l'équipement (carte
d'identité) ;
- La fiche de vie
permettant d'enregistrer les opération d'étalonnage,
de vérification, de contrôle, de calibrage et de
suivi de l'équipement ;
- Une fiche de
maintenance afin d'enregistrer les opérations de
maintenance en l'état (nettoyage, entretien,
...)
- Une fiche
d'intervention permettant d'enregistrer les opérations
suite à un dysfonctionnement.
3.6
Disposition des procédures et planification des instruments de
mesure (paragraphe 5.5.6)
Ce paragraphe ne
concerne que les instruments de mesure, la procédure de
maintenance y est imposée. Les autres procédures ne
devront exister que si elles présentent un
intérêt en terme de gestion d'un risque particulier (ex
: transport des équipements de mesure que si le
matériel est déplacé).
3.7
Réparation des défaillances des équipements
(paragraphe 5.5.7)
La norme impose un
étiquetage spécifique des équipements
présentant un dysfonctionnement. Elle rappelle qu'il est
nécessaire d'avoir une procédure afin de gérer
les non-conformités.
3.8
Codification des équipements du laboratoire (paragraphe
5.5.8)
Une étiquette
précisant la prochaine date d'étalonnage doit figurer
sur l'équipement afin que l'opérateur puisse identifier
si l'équipement qu'il va utiliser est conforme pour sa
périodicité d'étalonnage.
3.9
Vérification de l'état de fonctionnement de
l'équipement (paragraphe 5.5.9)
Cette exigence
s'applique sur un équipement mis à disposition de deux
sections dont l'une n'est pas soumise à l'accréditation
et qui se trouve régulièrement dans cette
position.
3.10
Vérification intermédiaire (paragraphe
5.5.10)
Si cela le
nécessite, il faut réaliser des vérification
intermédiaires (entre 2 étalonnages) en fonction des
risques liés à l'utilisation des équipements
(notamment au niveau de la fiabilité des mesures).
3.11
Disposition de procédures (paragraphe 5.5.11)
Les certificats
d'étalonnage devront être archivés et les
opérateurs pourront y avoir accès afin d'apporter
éventuellement des facteurs de correction.
3.12
Protection des équipements (paragraphe 5.5.12)
Cette exigence doit
s'appliquer au cas par cas, néanmoins pour certains
équipements il faudra mettre en place des modes
opératoires afin de garantir le bon réglage de
l'équipement pour qu'il accomplisse sa fonction.
4.
Traçabilité du mesurage
4.1
Généralités (paragraphe 5.6.1)
Tout
équipement de mesure devra être raccordable aux
étalons nationaux ou internationaux pertinents. Cependant, il
faut intégrer l'étalonnage des équipements qui
servent à surveiller les conditions ambiantes.
4.2
Prescriptions spécifiques : l'étalonnage (paragraphe
5.6.2)
Pour les laboratoires
d'essai, l'exigence est réduite aux seuls équipements
dont les fonctions de mesurage ont une incertitude qui a une
influence significative sur le résultat final.
4.3
Etalons de référence et matériaux de
référence (paragraphe 5.6.3)
Pour la
réalisation du test de sécurité
électrique, on n'utilise pas d'étalon pour calibrer nos
testeur. Le laboratoire ne sera donc pas concerné par ces
exigences. Elle pourra éventuellement être applicable
aux appareils qui mesure les conditions ambiantes.
5.
Planification des procédures de l'échantillonnage et
manutention des objets
5.1
Echantillonnage (paragraphe 5.7)
Dans le cadre d'un
laboratoire d'essai de la sécurité électrique on
ne réalisera pas d'échantillonnage.
5.2
Manutention des objets d'essai et d'étalonnage
5.2.1
Procédure de protection des objets d'essai et
d'étalonnage (paragraphe 5.8.1)
Le laboratoire doit
avoir des procédures pour le transport, la réception,
la manutention, la protection, le stockage, la conservation d'objets
à tester.
5.2.2
Identification des objets d'essai (paragraphe 5.8.2)
Le laboratoire doit
mettre en œuvre un système assurant l'identification du
dispositif à tester afin de garantir sa
traçabilité dans le laboratoire par exemple par la mise
en place d'un système à code barre.
5.2.3 Enregistrement des objets non conformes (paragraphe
5.8.3)
Cette exigence traite
ici de l'aspect technique et opérationnel de la revue de
contrat. En cas d'écart ou de doute, le laboratoire doit
procéder à un enregistrement et à une prise de
contact avec le client.
L'élaboration
d'un « dossier essai » permettant la vérification de
l'objet à la réception, puis l'enregistrement et la
gestion de l'objet jusqu'à son retour au client permet de
prendre en compte les enregistrements des paragraphes 5.6.1 à
5.6.3. Il convient de définir les critères techniques
d'acceptation de l'équipement à la réception et
de les enregistrer pour que le personnel chargé de cette
vérification puisse le faire selon des processus
clairs.
5.3
Procédures et installation (paragraphe 5.8.4)
Les critères
d'acceptation à la réception doivent non seulement
être satisfaisant, mais il doit également exister des
dispositions et des installations permettant de maintenir
l'intégrité de l'équipement dans tous le
laboratoire. Les étapes préalables à la
réalisation des essais doivent faire l'objet de toutes les
instructions pertinentes en prenant en compte les installations
existantes.
Les conditions de mise
en sécurité, lorsqu'elles sont
nécessaires, doivent être définies et mise en
œuvre.
6.
Surveillance de la qualité, des résultats d'essai et
rédaction des rapports.
6.1
Assurer la qualité des résultats d'essai et
d'étalonnage (paragraphe 5.9)
Pour
développer l'assurance qualité et le management de la
qualité, le laboratoire doit procéder à un
contrôle qualité, c'est à dire à une
vérification des résultats d'essai ou des processus du
laboratoire.
Le laboratoire doit donc
conduire une réflexion importante sur la nature des
contrôles qualité nécessaires à
l'obtention de la confiance sur les résultats obtenus. Il faut
donc définir le processus d'analyse et les
responsabilités organisationnelles et techniques capables de
décider du lancement des action correctives nécessaires
en cas d'écart.
6.2
Rapport sur les résultats (paragraphe 5.10.1 à
5.10.3)
Ce paragraphe
définit les différents éléments du
rapport d'essai. S'il s'agit d'essais ou d'étalonnages
effectués pour des clients internes ou s'il existe un accord
écrit avec le client, les résultats peuvent être
consignés de manière simplifiée.
6.3
Certificats d'étalonnage ( paragraphe 5.10.4)
Ce paragraphe apporte des
précisions pour les laboratoire qui délivrent des
certificats d'étalonnage, ce qui n'est pas le cas de notre
laboratoire d'essai.
6.4
Avis et interprétations (paragraphe 5.10.5)
Les avis et
interprétations sont autorisés avec certaines
limitations logiques : les bases sur lesquelles ils se reposent sont
formellement identifiées et écrites, et ils
apparaissent clairement différenciées des
résultats eux-mêmes.
6.5
Résultats d'essai et d'étalonnage obtenus auprès
de sous-traitants (paragraphe 5.10.6)
Lorsqu'un
étalonnage a été effectué par un
sous-traitant, le laboratoire qui a exécuté cette
tâche doit délivrer le certificat d'étalonnage au
laboratoire contractant.
Dans le cas de notre
laboratoire, il n'y a pas de sous-traitant, il ne faut donc pas tenir
compte de cette exigence
6.6
Présentation des résultats
6.6.1
Transmission électronique des résultats (paragraphe
5.10.7)
Pour respecter cette
exigence, il convient d'intégrer les recommandation de la
Société Française d'Informatique de
Laboratoire.
6.6.2
choix de la présentation du rapport (paragraphe
5.10.8)
Le rapport est le
produit du laboratoire. Pour le client, c'est ce qui reste de la
prestation. En plus de la qualité des résultats
fournis, le fond et la présentation du rapport, donnent une
image de la qualité du laboratoire. Ces deux
éléments doivent être pris en compte dans
l'élaboration des différents formulaires.
6.6.3 Amendements aux rapports d'essai et aux certificats
d'étalonnage (paragraphe 5.10.9)
Lorsqu'un rapport est
réémis dans sa totalité en raison d'une erreur
du laboratoire, il convient de s'interroger sur l'opportunité
d'obtenir le retour du premier original envoyé afin de ne pas
voir ce document utilisé de façon erronée. Sur
le deuxième document figure toutes les indications, notamment
l'annulation du précédent rapport et son remplacement
par celui-ci.
Cette demande de retour
a pour but de sensibiliser le client au profond changement qui
affecte le document. Cependant, ceci doit être fait avec la
plus grande diplomatie, le client pouvant imaginer que le laboratoire
met en doute son intégrité alors qu'il ne s'agit que de
l'aider à éviter des erreurs par l'utilisation non
intentionnelle de résultats erronés.
III
METHODE DE MISE EN PLACE DANS UN LABORATOIRE
BIOMEDICAL.
La partie qui va suivre est
donnée comme méthode pour la mise en place d'une
démarche qualité pour la certification d'un laboratoire
de tests de sécurité électrique à la
norme ISO 17025. Cette méthode prend donc ce cas comme exemple
mais celle-ci peut également être appliquées
à d'autres cas d'essais. Des précisions seront
régulièrement données afin d'assurer
l'éventuelle reproductibilité de cette méthode
dans un laboratoire biomédical.
Analyse des
éléments liés à la réalisation de
l'essai :
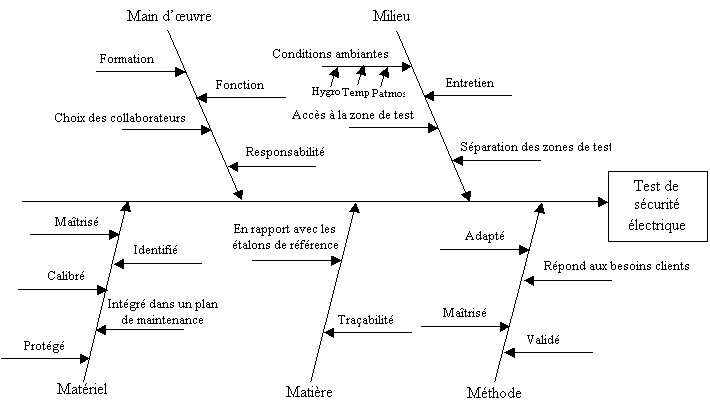
Figure 10 : diagramme
"causes effets" pour l'évaluation de la sécurité
électrique selon la norme ISO 17025
A)
PLANIFICATION DU PROJET EN VUE D'UN ACCREDITATION
17025
Que le projet de mise en
place d'un système qualité dans un laboratoire soit
imposé ou qu'il relève de la seule volonté de
son management, le responsable de projet devra avant tout
déterminer une stratégie générale en
matière de qualité à l'intérieur d'une
stratégie globale de l'entreprise, effaçant ainsi
l'impression de qualité subie par des contraintes. Le
responsable devra donc afficher quotidiennement sa motivation et sa
volonté afin de mener à bien ce projet.
Pour obtenir son
accréditation selon la norme 17025, le laboratoire devra
définir sa politique qualité, désigner son
responsable projet, sensibiliser et motiver son personnel,
déterminer l'architecture du système qualité,
définir les documents qualité et répartir les
tâches de rédaction.
Pour réaliser cette
partie, nous avons collaboré avec une équipe
d'étudiants du module « GE37 gestion de projet » de
l'UTC. Un rapport à été rédigé
rassemblant leurs méthodologies et conclusions.
[1]
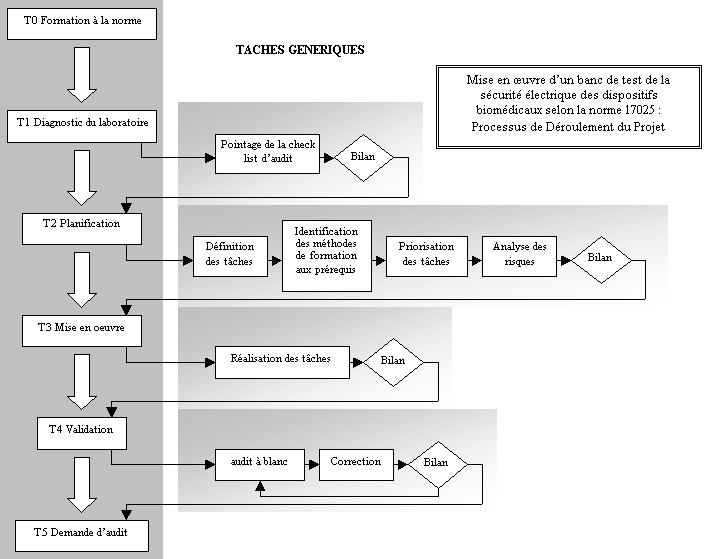
Les outils de gestion de
projet qui ont été utilisés sont :
- Le Processus de
Déroulement du Projet (PDP) ;
- Le questionnaire
d'audit selon la norme ISO 17025 ;
- Le listing des
tâches ;
- L'Analyse des Modes de
Défaillances, de leurs Effets et de leur Criticité
(AMDEC) ;
- Le tableau
d'identification des pré-requis et des formations
;
- Le tableau
d'identification et d'affectation des ressources ;
- Les plannings de Gantt
;
Le PDP a permis
d'identifier et de présenter l'enchaînement des 6
macro-tâches successives du projet. Chaque macro-tâche
est définie par des principales tâches
génériques, le but étant de préciser les
idées directrices.
La première
macro-tâche est T0 qui correspond à la formation
à la norme. En effet, pour pouvoir répondre aux
exigences de la norme 17025, il est important que tous les acteurs du
projet se forment à cette norme.
La seconde
macro-tâche est T1 qui correspond au diagnostic initial du
laboratoire. Réaliser l'état des lieux du
laboratoire à accréditer permet de pouvoir
évaluer le travail à fournir. Pour ce faire, la
tâche générique « pointage du questionnaire
d'audit » à partir de laquelle il est possible de
réaliser un premier bilan a été
définie.
La troisième
macro-tâche est T2 qui correspond à la
planification du projet. En sortie de cette macro-tâche, on
doit disposer du référentiel de gestion. Les
tâches génériques de définition des
tâches en accord avec les exigences de la norme 17025 y figure
puis d'identification des méthodes de formation aux pré
requis, d'analyse des risques et enfin de priorisation des
tâches.
La quatrième
macro-tâche est T3 correspond à la mise en œuvre
du projet. La tâche générique est donc la
réalisation des tâches définies en T2 et selon le
référentiel de gestion également défini
en T2.
La cinquième
macro-tâche est T4 qui correspond à la validation de
toutes les actions entreprises. La première tâche
générique est donc la réalisation d'un audit
à blanc grâce au questionnaire d'audit. A la suite, une
seconde tâche générique d'actions correctives
à entreprendre pour corriger les problèmes de non
conformité est à réaliser. A la suite de ces
actions correctives, un quatrième bilan est
réalisé. Si aucun problème ne subsiste alors on
peut passer à la dernière macro-tâche.
La sixième et
dernière macro-tâche est T5 qui correspond à
la demande d'Audit auprès du Comité
Français d'Accréditation : la COFRAC.
Ce Processus de
planification permet donc d'avoir une vision d'ensemble du projet et
c'est le point de départ essentiel pour planifier
précisément un projet.
B)
MACRO PROCESSUS ET CYCLES DE VIE
La méthode suivante
devrait permettre de structurer le système
qualité.
Les processus
permettront d'intoduire la méthode en définissant les
éléments entrants et sortants d'une activité.
Nous avons donc ici à faire à des macro processus qui
seront ensuite plus détaillés par d'autres
méthodes. Ces processus permettent donc une vision globale
pour mieux aborder l'organisation du manuel
qualité.
Le cycle de vie,
c'est l'ensemble des phases qui permettent de réaliser un
activité déterminée. Pour chacunes des phases,
on pourra examiner s'il existe ou non des dispositions
précises, pré établies voire formalisées
de l'organisation propre au laboratoire.
Dans le cas contraire,
on pourra à loisir compléter ces cycles s'ils semblent
trop dépouillés. Ici les cycles seront très
simplifiés puisque uniquement appliqués à un
laboratoire qui ne ferait que des tests de sécurité
électrique.
Le laboratoire doit prendre
en compte et expliciter le ou les domaine(s) d'activités qu'il
veut faire accréditer et les transposer dans un système
qualité. Pour ce faire, le responsable de la mise en place de
la démarche qualité devra analyser l'existant et mettre
en place toutes les réponses dictées par les exigences
de la norme. Afin de définir au mieux les points
concernés par la démarche qualité, une
méthode est proposée ci-dessous utilisant de nombreux
outils de qualité notamment les processsus et les cycles de
vie.
En effet, le système
qualité sera d'autant plus facile à gérer qu'il
sera soigneusement structuré.
La
représentation des cycles de vie est celle d'un cycle
fermé en sachant qu'on n'attend pas la fin d'un cycle pour
entamer le second. Les divers cycles sont inter dépendants les
uns des autres.
Nous allons tenter
d'expliciter les macro-processus à l'aide de cycles de vie
eux-même décomposés à l'aide de plusieurs
outils : diagrammes Ishikawa, processus, organigramme...
Ci-dessous le schéma
général donnant la trame de ce paragraphe:
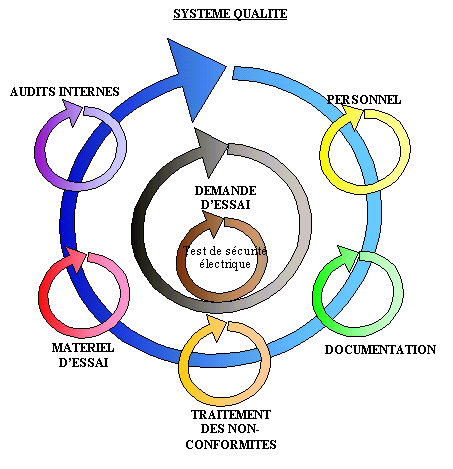
Figure 11 :
définition du système d'assurance
qualité
Il faudrait pour bien
détailler les activités pouvoir préciser tous
les processus associés à chaque étape de chaque
cycle de vie.
Ayant
décidés de ne détailler que les exigences
techniques de la norme dans cette partie, ne seront
détaillés que :
- Le cycle de vie de la
demande d'essai
- Le cycle de vie du test
de sécurité électrique
- Le cycle de vie du
matériel qui sert à effectuer les essais
- Le cycle de vie du
personnel
- a)
Réponses au exigences techniques.
1.
macro processus global : « évaluation de la
sécurité électrique selon la norme 17025
».
Il représente
l'activité du laboratoire d'essais d'une manière
globale.
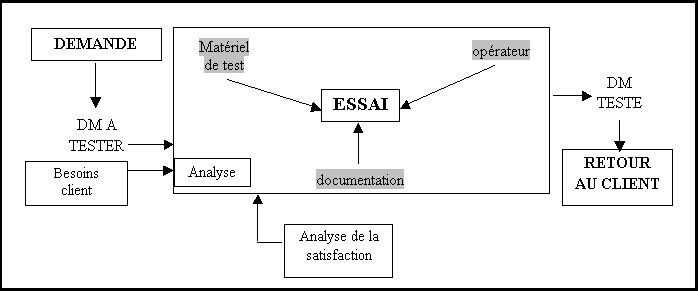
Figure 12 : macro
processus global représentant l'activité globale du
laboratoire .
Le cycle de vie de la
demande d'essai détaille le macro processus ci-dessus
:
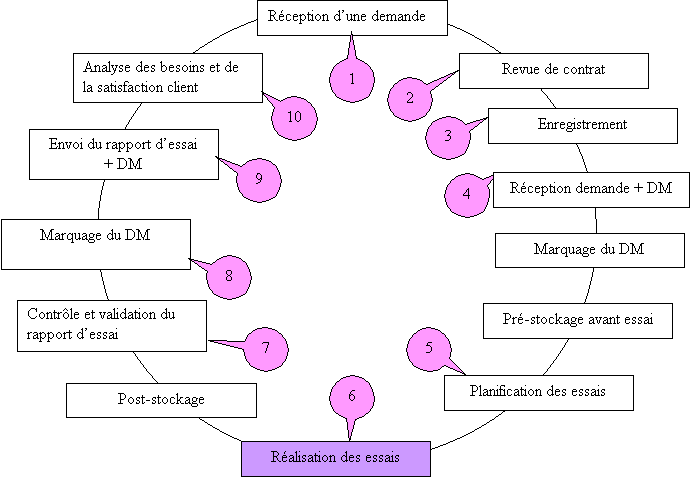
Figure 13 : cycle de
vie de la demande d'essai
|
ETAPE
DU CYCLE DE VIE
|
PARAGRAPHE
DE LA NORME
|
|
Réception
d'une demande
|
4.4.1 ;
4.4.3
|
|
Revue de
contrat
|
4.4.1
|
|
Enregistrement
|
4.4.2
|
|
Réception
demande + DM
|
5.8.4 ;
5.10.2 ; 5.10.3
|
|
Marquage
du DM
|
5.8.2 ;
5.8.3
|
|
Pré-stockage
avant essai
|
5.8.4
|
|
Planification
des essais
|
4.4.1
|
|
Réalisation
des essais
|
5.1.2
;5.3.1 ; 5.3.2 ; 5.3.3 ;5.3.4 ;5.3.5 ; 5.4.1 ;
5.10.1
|
|
Post-stockage
|
5.8.4
|
|
Contrôle
et validation du rapport d'essai
|
5.4.5 ;
5.4.6 ; 5.10.2 ; 5.10.3 ; 5.10.5 ; 5.10.8 ;
5.10.9
|
|
Marquage
du DM
|
5.8.2
|
|
Envoi du
rapport d'essai+ DM
|
5.4.7 ;
5.10.7
|
|
Analyse
des besoins et de la satisfaction client
|
4.7 ;
4.4.5
|
1. réception d'une demande.
|
Entrees
|
Contenu
|
Indicateurs
|
Sorties
|
|
Demande pour la
réalisation d'un essai.
|
Identification
de la demande client :
- demandeur
- dispositif
médical
- accessoires
et consommables
- dates
|
Taux de
formulaires correctement remplis.
Taux de champs
correctement remplit dans le formulaire
Retour
d'insatisfaction client.
|
Formulaire de
demande d'essai
|
2. Revue de contrat
|
Entrees
|
Contenu
|
Indicateurs
|
Sorties
|
|
Formulaire de
demande d'essai
|
Vérification
de l'adéquation besoins du client et prestations
fournies
|
- Nombre de
demandes sans suite
- Taux
d'insatisfaction client.
|
Devis
accepté
|
3. Enregistrement
|
Entrees
|
Contenu
|
Indicateurs
|
Sorties
|
|
Devis
accepté
|
Enregistrement
de la demande
|
- Taux des
éléments constitutifs du
dossier
- Delai
d'ouverture d'un dossier
|
Dossier
ìessaiî référencé pour
chaque demande.
|
4. Réception demande + DM
|
Entrees
|
Contenu
|
Indicateurs
|
Sorties
|
- Dossier
ìessaiî
- DM à
tester
- Bon de
réception
|
Réception
des documents et du DM
|
Délais
de réception
|
- Bon de
réception dans le dossier
ìessaiî
- DM à
tester marqué d'un code barre en
pré-stockage.
|
5. Planification
|
Entrees
|
Contenu
|
Indicateurs
|
Sorties
|
- Dossier
ìessaiî
- Planning
des essais
|
Planification
de l'essai
|
|
- Fiche de
panification dans le dossier
ìessaiî
- Planning
des essais mis à jour
|
6. Réalisation de l'essai
|
Entrees
|
Contenu
|
Indicateurs
|
Sorties
|
- DM à
tester
- Dossier
ìessaiî
|
Réalisation
physique des essais
|
Taux d'essais
correctement réalisés dans le temps imparti
sans problèmes de non conformté
|
- DM
testé mis en post stockage
- Fiche de
résultats dans le dossier
ìessaiî
|
La phase
réalisation des essais n'a pas été
détaillée. Elle fera l'objet d'un cycle de vie
« le test » dans le paragraphe suivant. En effet, cette
étape était a spécifier dans ce cycle
naturellement entre la demande d'essai et l'envoi du rapport
d'essai. Ces deux cycles sont directement liés l'un
à l'autre.
Figure 14 :
Agencement des cycles de vie du test et de la demande
d'essai.
7. Contrôle et validation du rapport
d'essai
|
Entrees
|
Contenu
|
Indicateurs
|
Sorties
|
- Dossier
ìessaiî
- Norme EN 60
601-1
|
Analyse et
validation des résultats donnés par les
testeurs
|
- Nombre de
rapport refusé
- Délai
de validation des résultats
|
Dossier
ìessaiî avec tampon de contrôle et
validation.
|
8. Marquage du DM
|
Entrees
|
Contenu
|
Indicateurs
|
Sorties
|
- DM +
étiquette
- Dossier
ìessaiî
|
Marquage du
résultat et de la date limite de validité
(selon exigences règlementaires) sur le
DM
|
Taux de DM non
marqués
|
- Dossier
ìessaiî
- DM
marqué
|
9. Renvoi du rapport d'essai + DM
|
Entrees
|
Contenu
|
Indicateurs
|
Sorties
|
- DM
testé et marqué
- Dossier
ìessaiî validé
|
Conditionnement
et renvoi
|
- Délai
de retour
- Taux
d'insatisfaction client.
|
- Bon de
livraison
- Double
exemplaire du dossier ìessaiî
|
10. Analyse des besoins et de la satisfaction
client
|
Entrees
|
Contenu
|
Indicateurs
|
Sorties
|
|
Retours
enquêtes et audits clients
|
Analyse des
besoins explicites, implicites et latents et de la
satisfaction des clients.
|
- Nombre
d'enquête annuelle
- Nombre de
retours clients
|
Dossier de
synthèse sur les améliorations à
envisager.
|
2.
Cycle de vie du test :
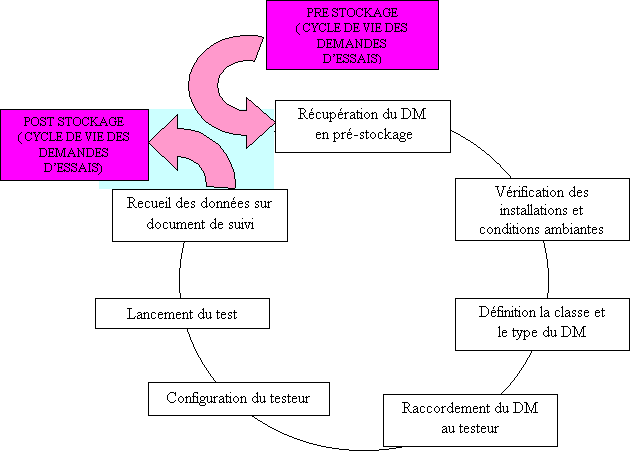
Figure 15 : cycle de
vie du test.
3.
Le processus matériel de tests:
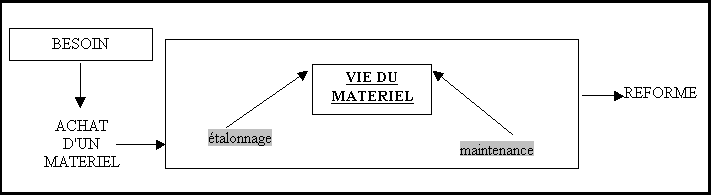
Figure 16 :
macro-processus du matériel
d'essais
Le cycle de vie «
matériel d'essai » c'est à dire le
matériel utilisé pour effectuer l'essai
détaille le macro- processus ci-dessus :
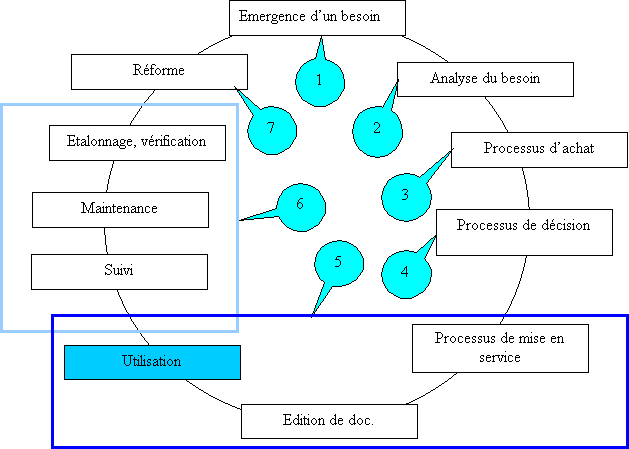
Figure 17 : cycle de
vie du matériel d'essai
|
ETAPE
DU CYCLE DE VIE
|
PARAGRAPHE
DE LA NORME
|
|
Emergence
d'un besoin
|
5.5.1
|
|
Analyse du
besoin
|
5.5.1
|
|
Processus
d'achat
|
4.6.1
|
|
Processus
de décision
|
4.6.1
|
|
Processus
de mise en service
|
4.6.2 ;
5.5.4
|
|
Edition de
doc
|
4.6.3 ;
5.5.5 ; 5.5.8 , 5.5.11
|
|
Utilisation
|
5.5.3
|
|
Suivi
|
5.5.2 ;
5.5.6
|
|
Maintenance
|
5.5.7 ;
5.5.12
|
|
Etalonnage,
vérification
|
5.5.9 ;
5.5.10
|
|
Réforme
|
5.5.1
|
3.1 Emergence d'un besoin
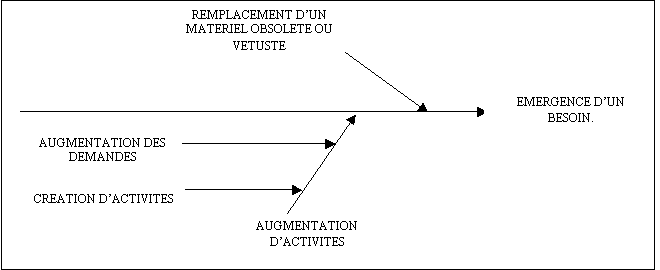
3.2 Analyse du besoin
|
Entrees
|
Contenu
|
Indicateurs
|
Sorties
|
- Demande des
clients
- Rapport
d'activité
- Etat du
parc
|
Identification
des besoins par rapport aux clients et au parc
existant.
|
- Nombre de
demandes
- Nombre
d'appareils en exploitation
|
- Document de
synthèse
- Document de
demande d'achat.
|
3.3 Processus d'achat
|
Entrees
|
Contenu
|
Indicateurs
|
Sorties
|
- Document
de synthèse
- Document de
demande d'achat
- Normes
règlementaires.
|
Identification
du matériel à acheter -
|
- Satisfaction
du demandeur
- Offre du
marché
|
Cahier des
charges.
|
3.4 Processus de décision
|
Entrees
|
Contenu
|
Indicateurs
|
Sorties
|
- Cahier des
charges
- Différentes
offres
|
Identification
du fournisseurs à retenir
|
Satisfaction du
demandeur
|
Bon de
commande
|
3.5 Réception et mise en service
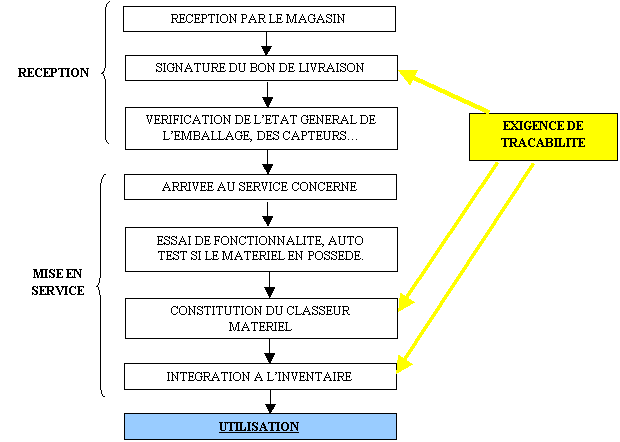
Figure 18 :
Organigramme de la réception et la mise en service du
matériel d'essai
Une fois de plus,
l'utilisation du matériel est spécifié mais
n'est pas détaillée. Pour ce faire, il faudra se
référer au paragraphe « cycle de vie du test
».
3.6 Suivi, maintenance et étalonnage d'un
équipement.
Dans ce cas, on
considère que la maintenance corrective et la maintenance
préventive sont faites chez le constructeur et que pour
l'étalonnage, on envoie le testeur dans un laboratoire
d'étalonnage habilité par un étalon national
ou international qui l'effectuera dans les conditions optimales
demandées par la norme associée
(NB : attention, le
fabricant ne l'est pas toujours).
3.6.1 Maintenance corrective.
|
Entrees
|
Contenu
|
Indicateurs
|
Sorties
|
- Testeur en
panne ou testeur défaillant pour une
fonctionnalité
- Fiche de
demande d'intervention
|
Envoi du
testeur au constructeur et identification du coût
de la réparation
|
- Temps de
retour de la défaillance
- Auto-test
|
- Testeur
dépanné
- Fiche
d'intervention
- Bon de
livraison
|
3.6.2 Maintenance préventive.
|
Entrees
|
Contenu
|
Indicateurs
|
Sorties
|
- Testeur
soumis à maintenance préventive selon
une période
déterminée
- Demande
d'intervention
|
Envoi du
testeur au constructeur et identification du coût
de la maintenance
|
- Nombre de
maintenance corrective
- Auto-test
|
- Testeur
testé
- Fiche
d'intervention
- Bon de
livraison
|
3.6.3 Etalonnage.
|
Entrees
|
Contenu
|
Indicateurs
|
Sorties
|
-
- Testeur
soumis à étalonnage selon une
période déterminée
- Demande
d'intervention
|
Envoi du
testeur à un organisme habilité à
effectuer un étalonnage
adéquat
|
Cohérence
des résultats obtenus aux tests de
sécurité électrique
|
- Testeur
étalonné
- Fiche
d'intervention
- Bon de
livraison
|
3.7 Réforme
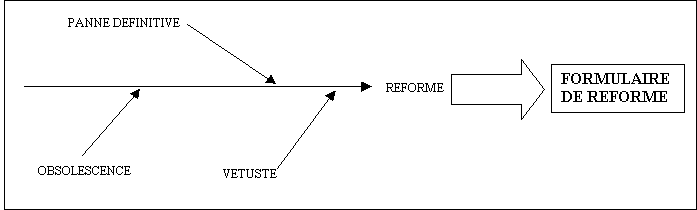
- 4.
Le processus personnel:
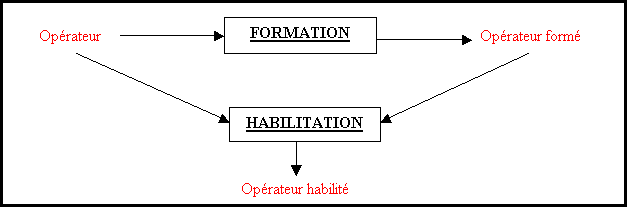
Figure 19 :
macro-processus de la gestion du personnel.
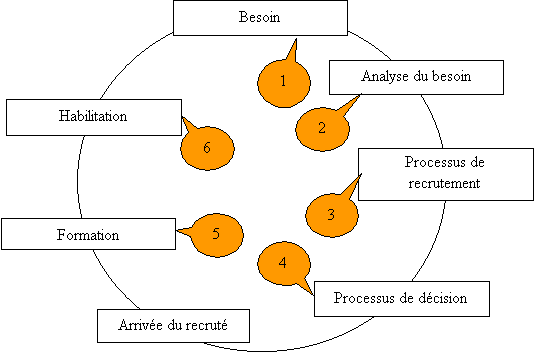
Figure 20 : cycle de
vie du personnel
Le personnel doit
être habilité à effectuer le test à la
date du test. Dans ce cycle, l'étape « formation
» correspond d'une part à la formation technique sur
les pratiques internes, et d'autre part à la formation sur
le système qualité mis en œuvre au sein du
laboratoire.
L'habilitation sera
donnée par un supérieur hiérarchique pour une
durée déterminée ou
indéterminée.
|
ETAPE
DU CYCLE DE VIE
|
PARAGRAPHE
DE LA NORME
|
|
Besoin
|
5.2.4
|
|
Analyse du
besoin
|
5.2.3
|
|
Processus
de recrutement
|
5.2.4
|
|
Processus
de décision
|
5.2.4
|
|
Arrivée
du recruté
|
5.2.4
|
|
Formation
|
5.2.1 ;
5.2.2
|
|
Habilitation
|
5.2.4 ;
5.2.5
|
4.1 Besoin.
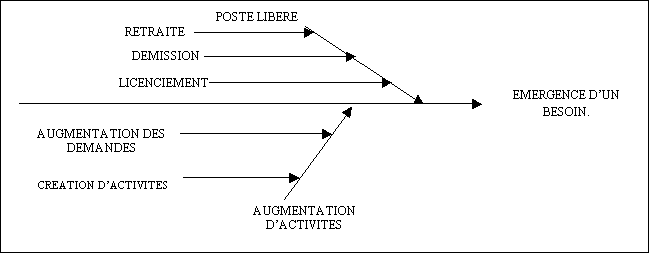
4.2 Analyse du besoin
|
Entrees
|
Contenu
|
Indicateurs
|
Sorties
|
- Demande
- Rapport
d'activité
- Analyse du
personnel
|
Identification
des besoins par rapport aux clients et à
l'activité
|
- Nombre de
demande
- Nombre
d'employés
|
Document de
synthèse + définition du poste (fiche de
poste)
|
4.3 Processus de recrutement
|
Entrees
|
Contenu
|
Indicateurs
|
Sorties
|
-
- Dossier
employé ( cv, lettre de motivation, photo
...)
- Document de
synthèse
|
Identification
du profil recherché
|
- Profil de
l'ancien employé
- Compétences
par rapport aux pratiques
|
- Définition
du profil
- Préparation
des entretiens
|
4.4 Processus de décision
|
Entrees
|
Contenu
|
Indicateurs
|
Sorties
|
- Entretien-
Dossier employé ( cv, lettre de motivation,
photo ...)
- Document de
synthèse
|
Identification
du candidat à retenir
|
Adéquation
définition du profil / candidat
retenu
|
- Un candidat
unique
- Un contrat
de travail
- Un dossier
employé
|
4.5 Formation
|
Entrees
|
Contenu
|
Indicateurs
|
Sorties
|
|
Dossier
ìemployéî
|
Formation
technique et/ou qualité
|
Contrôle
des connaissances
|
Dossier
ìemployéî + attestations de
formations internes
|
4.6 Habilitation.
|
Entrees
|
Contenu
|
Indicateurs
|
Sorties
|
- Formation
- Savoir
faire + personnalité
- Dossier
ìemployéî
|
Identification
de l'adéquation compétences /
activités
|
- Satisfaction
du reste du personnel
- Satisfaction
du client.
|
Dossier
ìemployéî + fiche
d'habilitation.
|
Le dossier «
employé » contiendra donc :
- Le CV
- Les photocopies des
diplômes
- Résumés
des rapports de stages, thèses,
publications,
- Les attestations de
formations internes
- La fiche de
poste
- La fiche
d'habilitation.
b)
Réponses aux exigences organisationnelles.
1.
Le processus documentation:
-
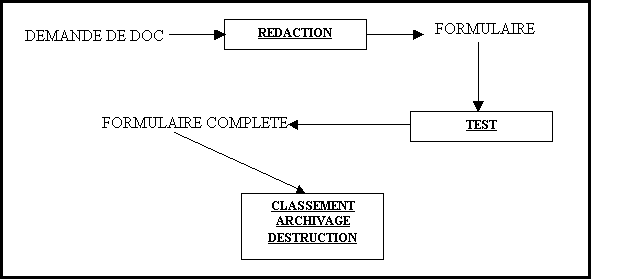
Figure 21 :
macro-processus de la documentation
Le cycle de vie de la
documentation détaille le macro processus ci
dessus.
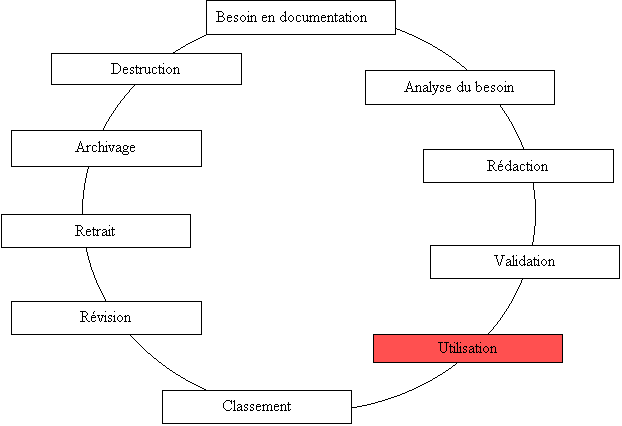
Figure 22 : cycle de
vie de la documentation
|
ETAPE
DU CYCLE DE VIE
|
PARAGRAPHE
DE LA NORME
|
|
Besoin en
documentation
|
4.3
|
|
Analyse du
besoin
|
4.3
|
|
Rédaction
|
4.3
|
|
Validation
|
4.3.2
|
|
Utilisation
|
4.12.1
|
|
Révision
|
4.3.3
|
|
Retrait
|
4.3.2
|
|
Archivage
|
4.12.1
|
2.
Le processus de traitement des non
conformités.
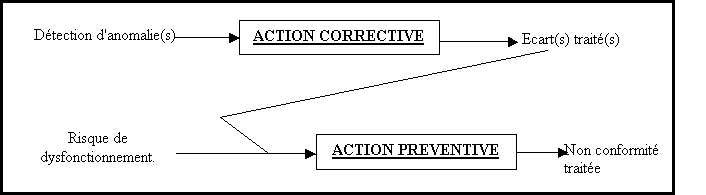
Figure 23 :
macro-processus de la gestion des
anomalies.
Le cycle de vie de
la gestion des non conformités détaille le macro
processus ci dessus.
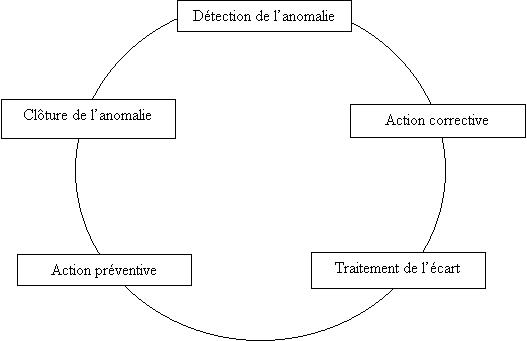
Figure 24 : cycle de
vie de la gestion des anomalies
|
ETAPE
DU CYCLE DE VIE
|
PARAGRAPHE
DE LA NORME
|
|
Détection
de l'anomalie
|
4.9.1
|
|
Action
corrective
|
4.10.3
|
|
Traitement
de l'écart
|
4.10.4 ;
4.10.5
|
|
Action
préventive
|
4.11.1 ;
4.11.2
|
|
Clôture
de l'anomalie
|
4.9.1
|
3.
Le processus des audits internes.

Figure 25 :
macro-processus de l'audit interne.
Le cycle de vie des
audits internes détaille le macro processus ci
dessus.
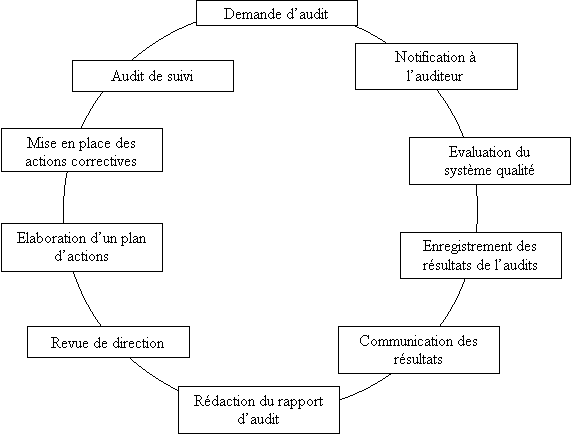
Figure 26 : cycle de
vie de l'audit interne
|
ETAPE
DU CYCLE DE VIE
|
PARAGRAPHE
DE LA NORME
|
|
Demande
d'audit
|
4.13.1
|
|
Notification
à l'auditeur
|
4.13.1
|
|
Evaluation
du système qualité
|
4.14.2
|
|
Enregistrement
des résultats de l'audits
|
4.13.2
|
|
Communication
des résultats
|
4.13.2
|
|
Rédaction
du rapport d'audit
|
4.13.3
|
|
Revue de
direction
|
4.14.1
|
|
Elaboration
d'un plan d'actions
|
4.14.1
|
|
Mise en
place des actions correctives
|
4.13.4
|
|
Audit de
suivi
|
4.13.4
|
-
C)
METHODOLOGIE DE REDACTION
a) Le manuel qualité :
Le manuel
qualité est le pivot documentaire de tout le
système qualité.
Le manuel
qualité est définit par la norme ISO 8402 comme
:
« un document
énonçant la politique qualité et
décrivant le système qualité d'un
organisme.
Note 1 : un manuel
qualité peut porter sur la totalité des
activités d'un organisme ou seulement une partie de
celle-ci. Le titre et l'objet du manuel explicitent le champ
d'application.
Note 2 : un manuel
qualité contiendra normalement, ou fera
référence au moins :
La politique
qualité,
Les
responsabilités, les pouvoirs et les relations entre
personnes qui dirigent, effectuent, vérifient ou passent
en revue les travaux qui ont une incidence sur la
qualité.
Les procédures
et les instructions du système
qualité,
Des dispositions pour
revoir, mettre à jour et gérer le manuel
qualité.
Note 3 : pour
s'adapter aux besoins d'un organisme, le degrés de
détails et la forme d'un manuel qualité peuvent
varier. Le manuel peut être constitué de plusieurs
volumes. Quand le manuel traite des besoins de l'assurance de
la qualité, il est
parfois appelé
« manuel d'assurance qualité »
»
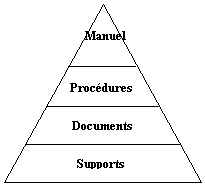
Le manuel qualité
fera référence aux procédures,
instructions et enregistrements, ce qui lui donne l'avantage
d'être :
- Facilement
consultable,
- Diffusable
à l'extérieur du laboratoire,
- Accessible.
Les
détails techniques sont donnés par les trois
étages inférieurs de la pyramide :
procédures, instructions et enregistrements.
Le manuel
qualité est strictement organisationnel et exclut toutes
descriptions techniques. Il a pour objectif d'apporter la
preuve que le laboratoire dispose de tous les moyens permettant
de satisfaire chaque exigence de la norme ISO 17025.
Ainsi
à partir de ce modèle pyramidal, nous avons
identifier deux manières d'aborder la documentation du
système qualité :
- A partir de la
méthode PIEM explicitée dans le §
II. Le manuel sera construit à partir des chapitres
de la norme et des exigences qu'elle contient.
- A partir des
cycles de vie explicités dans le § III B.
Chaque cycle de vie fera l'objet d'un chapitre du manuel
qualité. Les exigences de la norme devront être
judicieusement replacées.
Il paraît clair
que la deuxième approche semble plus laborieuse à
mettre en place mais elle sera pourtant plus facile à
gérer par la suite.
Pour les deux méthodes, on doit remplir les chapitres par
la manière dont on répond aux exigences dans le
laboratoire.
-
|
EXEMPLE
D'APPLICATION POUR LES EXIGENCES RATTACHEES AUX MATERIELS
D'ESSAIS
|
Cet
exemple regroupe les différentes exigences
rattachées aux matériels d'essais. On constate que
ces deux chapitres concernés ne sont pas successifs dans le
manuel qualité.
PARAGRAPHE
4.6 : ACHATS DE SERVICES ET RELATION AVEC LE
CLIENT
Procédure
d'achat
Politique d'achat de service et fournitures : M
Achat de services et fournitures : P et E
Achat, réception et stockage des réactifs et
fournitures : P et E
Produits
et services achetés
Contrôle ou vérification des fournitures avant
utilisation : M
Conformité des services et fournitures aux exigences
spécifiques : M
Dispositions de contrôle de conformité : I et
E
Contenu
des documents d'achats
Contenu des documents d'achat : M
Revue et approbation des documents d'achat : I et E
Evaluation
des fournisseurs de produits consommables, fournitures et services
critiques
Détermination de la criticité des fournitures et
services : I et E
Evaluation des fournisseurs : I et E
Rapport d'évaluation : E
Liste des fournisseurs approuvés : E
PARAGRAPHE
5.5 : ÉQUIPEMENT
Pour
un bon usage de l'équipement
Equipement du laboratoire : E
Utilisation d'un équipement non contrôlé en
permanence : I et E
Vérification
de l'équipement
Obtention de l'exactitude requise : M et E
Programme d'étalonnage : E
Etalonnage avant mise en service : I et E
Contrôle ou étalonnage avant utilisation : I et
E
Liens
entre le personnel et l'usage de
l'équipement
Qualification du personnel sur un matériel : I et
E
Modalité d'utilisation des équipements :
I
Modalités d'entretien des équipements : I et
E
Identification
des équipements
Identification unique des équipements : I et E
Identification
des équipements
Fiche de vie : E
Disposition
des procédures et planification des instruments de
mesure
Manutention des instruments de mesure : P et E
Transport des instruments de mesure : P et E
Utilisation des instruments de mesure : P et E
Maintenance planifiée des instruments de mesure : P et
E
Réparation
des défaillances des équipements
Mise hors service d'un équipement défectueux : I et
E
Isolement, marquage ou étiquetage avant remise en service :
I et E
Examen de l'effet de la défaillance : I et E
Lancement de la procédure de non-conformité : I et
E
Codification
des équipement du laboratoire
Marquage complet du statut d'étalonnage : I et E
Vérification
de l'état de fonctionnement de
l'équipement
Remise en service d'un équipement ayant
échappé au contrôle du laboratoire : I et
E
Vérification
intermédiaire
Vérifications intermédiaires : P et E
Périodicité des vérifications
intermédiaires : E
Disposition
de procédure
Mise à jour des corrections d'étalonnage : I et
E
Protection
des équipements
Protection contre les déréglages : I et
E
2.
Deuxième approche : la méthode des cycles de
vie.
On trouve des exigences sur
un même thème à divers endroits de la norme.
C'est pour cela que cette méthode sera plus difficile pour la
construction du manuel qualité mais plus facile pour sa
gestion. En effet, on regroupe sous une même rubrique les
différentes exigences relatives à un même
sujet.
La découpe la plus
accessible par rapport aux cycles de vie mis en place et par rapport
au fonctionnement du laboratoire d'essai est la suivante :
- Les moyens :
personnel, matériels, locaux, consommables.
- La mise en
œuvre de ces moyens : le test de sécurité
électrique, le traitement des demandes d'essai.
- La gestion de cette
organisation : la documentation, le traitement des
non-conformités, les audits internes.
Le tout est
précédé d'un présentation du laboratoire,
d'une description de la gestion de ce manuel qualité et
éventuellement d'un explication des terminologies et
abréviations utilisées.
Le sommaire pourrait donc
être ici :
GENERALITES :
Sommaire
Présentation du laboratoire
Gestion du manuel qualité
Abréviations et terminologie
REPONSE A LA NORME :
Politique et objectifs qualité
Objet et domaines d'applications
Les locaux

Le
personnel
Le
matériel
Les demandes
d'essai
Le test de
sécurité électrique
La
documentation
Le traitement
des non-conformités
Les audits
internes
|
EXEMPLE
D'APPLICATION POUR LES EXIGENCES RATTACHEES AUX MATERIELS
D'ESSAIS
|
LES
MATERIELS D'ESSAI
1.
CATEGORIE DE MATERIELS
Matériels d'essais
On
décrira ici quels sont les matériels d'essais:
testeurs, multimètres, oscilloscopes...que l'on trouve au sein
du laboratoire, quelles sont les meilleures conditions pour les
utiliser et comment sont effectuées les mesures des conditions
ambiantes.
Matériels accessoires
On
décrira ici quels sont les matériels accessoires pour
le raccordement spécifique des différents dispositifs
médicaux aux matériels de mesure.
2.
ACHAT, RECEPTION ET MISE EN SERVICE D'UN EQUIPEMENT
On
décrira dans ce paragraphe les étapes d'achat,
réception et mise en service de
l'équipement.
Processus d'achat
On
décrira en plus dans ce paragraphe la politique d'achat des
matériels de tests et le contenu des documents
d'achat.
Ce paragraphe
devra faire référence à :
- Une
procédure d'achat et une procédure de
stockage associées à des
enregistrements.
- Une
instruction pour la revue et l'approbation des documents
d'achat, une instruction pour la détermination de la
criticité du matériel acheté et une
instruction pour l'évaluation des fournisseurs
associées à des enregistrements.
- Un
formulaire pour lister les fournisseurs
approuvés.
Réception et mise en service d'un
équipement
On
décrira en plus dans ce paragraphe le contrôle et la
vérification des matériels d'essais avant utilisation
et leurs conformités aux exigences
spécifiques.
Ce paragraphe
devra faire référence à :
- Une
instruction pour les dispositions de contrôle de
conformité, une instruction pour l'identification unique
des équipements et une instruction pour l'étalonnage
avant la mise en service du matériel associées
à des enregistrements.
3.
GESTION DU PARC DEQUIPEMENT
Dossier matériel
C'est le
dossier du matériel de tests qui sera associé à
tous les enregistrements qui lui sont relatifs : fiche
signalétique de l'équipement, fiche de vie, fiche de
maintenance, fiche d'intervention.
Ce paragraphe
devra faire référence à :
- Une
procédure pour la maintenance planifiée des
matériels d'essais associée à des
enregistrements.
- Une
instruction sur l'entretien des matériels de tests,
une instruction pour la mise hors service d'un équipement
défectueux, une instruction pour l'isolement, le marquage
et l'étiquetage avant remise en service et une instruction
pour la remise en service d'un équipement ayant
échappé au contrôle du laboratoire
associées à des enregistrements.
Utilisations
On
décrira ici ou on fera référence à tout
ce qui est relatif à l'utilisation du matériel c'est
à dire tous les modes opératoires techniques mais aussi
toutes les activités latentes du laboratoire comme la
manutention ou le transport.
Ce paragraphe
devra faire référence à :
- Une
procédure sur la manutention, une procédure
pour le transport et une procédure pour l'utilisation des
matériels d'essais associées à des
enregistrements.
- Une
instruction sur la qualification du personnel qui travaille
sur le matériel d'essais, une instruction pour
l'utilisation des équipements, et une instruction sur le
contrôle avant utilisation associées à des
enregistrements.
4.
ETALONNAGE ET VERIFICATION
Etalonnage et vérification du matériels de
mesure
On
décrira ici les obligations d'étalonnage, les
paramètres à étalonner et l'exactitude requise
pour les résultats ou les délais à appliquer en
fonction du matériel d'essais concerné selon les
prescriptions fournisseurs et les référentiels
réglementaires. On décrira également les
vérifications intermédiaires.
Ce paragraphe
devra faire référence à :
- Une
procédure de vérifications
intermédiaires associée à des
enregistrements.
- Une
instruction sur le marquage complet du statut
d'étalonnage, une instruction sur la mise à jour des
corrections d'étalonnages et une instruction sur la
protection des déréglages, associées à
des enregistrements.
- Un
formulaire pour la saisie des programmes
d'étalonnage et un formulaire pour vérifier la
périodicité des vérifications
intermédiaires.
Vérification du matériels accessoires
Ce paragraphe
concerne le matériel accessoire qui devra être
vérifié régulièrement selon un
délai donné (Par exemple : vérification de
l'intégrité des câbles).
Ce paragraphe
devra faire référence à :
- Une
procédure de vérifications
intermédiaires associée à des
enregistrements.
- Un
formulaire pour vérifier la
périodicité des vérifications
intermédiaires.
-
b) Les
procédures
Une procédure est
définit par la norme ISO 8402 comme :
« une
manière spécifiée d'accomplir une
activité :
Note 1 : dans de
nombreux cas, les procédures sont exprimées par des
documents,
Note 2 : lorsqu'une
procédure est exprimée par un documents, il est
préférable d'utiliser le terme «
procédure écrite »,
Note 3 : une
procédure comprend généralement : l'objet et
le domaine d'application d'une activité, ce qui doit
être fait et qui doit le faire, quand, où et comment
cela doit être fait, quels matériels,
équipements et documents doivent être
utilisés, comment cela doit être
maîtrisé et enregistré »
Il est bien
précisé dans cette définition qu'une
procédure doit décrire les « matériels,
équipements et documents qui doivent être
utilisés » et non la manière dont ces
matériels sont utilisées. La procédure reste
donc un document strictement organisationnel même si elle
constitue un degrés de détail supplémentaire
par rapport aux grandes lignes du manuel
qualité.
Une procédure
prend la forme d'un logigramme qui enchaîne des
étapes et des décisions afin de passer des
données d'entrée aux données de sortie. A
chacune de ces phases, on associe un responsable (R) ou un
décideur pour chaque processus décisionnel (D) et
éventuellement celui qui exécute (E), celui qui
possède l'autorité (A), celui qui conseille (C)
et/ou celui qu'il faut informer ( I ).
Pour toutes les
procédures, les symboles devront être
homogènes.

le recto de la
procédure comporte complémentairement au logigramme
:
- Un titre de la
procédure ;
- Un objet et un
domaine d'application qui explicitent le pourquoi de la
procédure ;
- Les
abréviations utilisées dans le logigramme tant au
niveau des fonctions concernées qu'au différents
types de responsabilité assumée ;
- Des informations qui
viennent compléter les éléments contenus
dans le logigramme.
Liste des procédures
demandées par la norme : voir annexe 4
Liste des procédures
demandées par la méthode PIEM : voir annexe
2
|
EXEMPLE
D'APPLICATION POUR LES EXIGENCES RATTACHEES AUX MATERIELS
D'ESSAIS
|

c)
Les instructions :
Ce sont des documents
opérationnels techniques. La rédaction d'une
instruction n'est nécessaire que si le texte de
référence n'est pas suffisamment bien
explicité.
Si l'instruction est
nécessaire, il convient de la rédiger à l'aide
d'une méthode identique à celle qui est utilisée
pour une procédure, à la différence près
qu'une instruction est réalisée par une seule fonction
(pas d'interface organisationnelle sinon c'est une procédure)
et que le déroulement des actions est dans la majorité
des cas linéaire, c'est à dire sans choix.
Pour l'ISO 17 025, quatre
types de modes opératoires sont nécessaires
:
|
Modes
opératoires d'analyses et
d'essais
|
DOCUMENT
OPERATIONNEL TECHNIQUE
|
|
Modes
opératoires de
métrologie
|
DOCUMENT
OPERATIONNEL TECHNIQUE
|
|
Modes
opératoires d'utilisation, entretien, maintenance de
matériel
|
DOCUMENT
OPERATIONNEL TECHNIQUE
|
|
Instructions
administratives
|
DOCUMENT
OPERATIONNEL ORGANISATIONNEL
|
Une instruction technique
devra donc contenir :
- Titre, objet et
domaines d'application
- Références
normatives
- Principe de la
méthode : DM testé, paramètres et grandeurs
à mesurer, étendue
- Equipement
utilisé
- Conditions
d'environnement
- Limites d'utilisation
de la méthode
- Processus
opératoire avec vérifications préalables et
enregistrement des résultats
- Précautions de
sécurité
- Traitement des
données brutes
- Evaluation de
l'incertitude
- Présentation des
résultats
- Eventuels écarts
par rapport au texte de référence et
justifications.
d)
Les enregistrements :
Les enregistrements sont
réalisés sur des formulaires prédéfinis.
Il existe deux types de formulaires : des supports pour un seul
enregistrement ou des supports pour plusieurs
enregistrements.
Ces formulaires devront
faire l'objet d'une codification spécifique, qui devra
être décrite dans le manuel qualité. Ils doivent
être faciles à remplir, donc clairs et précis
dans leur présentation.
|
EXEMPLE
D'APPLICATION POUR LES EXIGENCES RATTACHEES AUX MATERIELS
D'ESSAIS
|


IV)
PERSPECTIVES
L'accréditation est
donc la reconnaissance formelle de la compétence technique
d'un organisme à effectuer une prestation concrète,
définie dans le domaine accrédité.
|
Personnel
|
Infrastructure
technique
|
Structure
organisatrice
|
- Suffisant et
qualifié
- Expérience
pratique dans le domaine technique
concerné
- Formation
continue
|
- Locaux
- Equipements
- Procédures
|
- Indépendance
- Impartialité
- Gestion de la
qualité
|
Par conséquent,
l'accréditation est un moyen d'instaurer la confiance,
permettant aux autorités, à l'industrie et à la
société de juger, si les laboratoire d'essais,
exécutent des tâches définies avec un haut niveau
de fiabilité.
Le seul
référentiel relatif au contrôle de qualité
en matière d'accréditation est la norme
européenne ISO/CEI 17025, qui est spécifique aux
laboratoires d'essais.
Si un service
biomédical décide de se faire accréditer EN
ISO/CEI 17025, il devra alors prouver :
- qu'il gère un
système de management de la qualité,
- qu'il est techniquement
compétent,
- qu'il s'appuie sur des
référentiels (exemple : sécurité
électrique EN NF 60601-1),
- qu'il est capable de
produire des résultats techniquement valables et
mesurables.
Il est donc possible
d'envisager un atelier biomédical certifié ISO 9001
pour tout ou une partie de ses activités, et
accrédité ISO 17 025 pour les contrôles
qualité. En effet, on retrouve beaucoup de corrélations
entre ces 2 normes, notamment sur l'organisation du système
qualité (voir annexe 4).
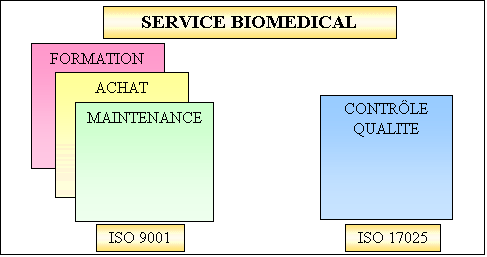
Cependant, le service
biomédical ne doit pas être juge et parti pour les
contrôles de qualité de ses dispositifs médicaux,
ceci étant un point délicat pour sa mise en application
au sein des services biomédicaux.
CONCLUSION
La certification ISO 9000
permet aux entreprises d'apporter à leurs clients la preuve de
la qualité de leur organisation.
Pour les laboratoires
d'analyses et d'essais, il est possible d'obtenir cette même
reconnaissance mais il est clair que l'aptitude technique du
laboratoire à réaliser les essais définis selon
des méthodes reconnues est d'une importance capitale.
L'accréditation ISO 17025, décernée par le
COFRAC, permet d'attester que les essais sont effectués
suivant des procédures et modes opératoires
référencés dans le système qualité
de l'organisme, seuls garants de leur
répétitivité et de la pérennisation des
compétences internes. Cette accréditation en plus
d'assurer la qualité de l'organisation du système,
garantie les résultats donnés par celle-ci.
Ce rapport est porteur d'une
amorce de mise en place de démarche qualité, pour un
laboratoire biomédical effectuant des tests de
sécurité électrique sur des dispositifs
médicaux. Dans une première partie, les exigences de la
norme ISO 17025 sont explicitées dans le cadre de ce
laboratoire. Dans une deuxième partie, une méthodologie
de mise en place sera proposée à l'aide d'une
décomposition des activités sous forme de processus
complétée par des points de repère pour la
construction du manuel qualité, des procédures, des
instructions et des enregistrements associés.
BIBLIOGRAPHIE
[1]
BOISDE C., CASSARO J., DEROUBAIX A., MINARET G., SENECHAL J., «
mise en œuvre d'un banc d'un équipement de laboratoire,
selon la norme ISO 17 025 », 2001-2002, module GE 37,
UTC.
[2] DESGIPPES
D., GALLOFRE J., GANDON M., « La sécurité
électrique des dispositifs électromédicaux
», 1999-2000, rapport
SPIBH,
UTC.
[3] FARGES
G., « sécurité électrique des dispositifs
médicaux », cours de juillet 2001.
[4] FARGES
G., LABARRE A., LORIMIER A., TAUPIAC S., « Contrôle
qualité et NF EN 45 001 : du service biomédical au
laboratoire biomédical », février 2001, vol 22
n°1, p6-8, RBM NEWS.
[5] NF EN 45
001, « Critères généraux concernant le
fonctionnement des laboratoires d'essais », décembre
1989, éditions AFNOR.
[6] NF EN 60
601-1, « Appareils électromédicaux : règles
générales de sécurité », novembre
1990, éditions AFNOR.
[7] NF EN
ISO/CEI 17 025, « Prescriptions générales
concernant la compétences des laboratoires
d'étalonnages et d'essais », mai 2000, éditions
AFNOR.
[8] REVOIL
G., « Assurance qualité dans les laboratoires d'analyses
et d'essais », éditions AFNOR,
1995.
[9] REVOIL
G., « Qualité dans les laboratoires d'analyses et
d'essais », éditions AFNOR,
2001.
ANNEXES
LE TEST
DE SÉCURITÉ ÉLECTRIQUE
ANALYSE
DES EXIGENCES DE LA NORME 17025
DÉFINITION
DES OUTILS DE LA PLANIFICATION DE
PROJET ET
DÉFINITION DES MACROTACHES
PROCEDURES
DEMANDEES PAR LA NORME
COMPARATIF
ISO 9001 ET ISO 17025
ANNEXE
1
LE TEST DE
SÉCURITÉ ÉLECTRIQUE
La norme NF EN 60 601-1
détermine les règles générales de la
sécurité électrique des équipements
médicaux. Elle définit les courants de fuite et
courants auxiliaires patient à mesurer. Les valeurs limites de
ces courants sont fixées selon la classification des
appareils.
Bon nombre d'entre eux font
l'objet de normes spécifiques qui modifient ou
complètent la norme générale.
L'étude combinée
de ces normes permet de développer des procédures de
contrôle de sécurité électrique. Celles-ci
regroupent l'identification de l'équipement, la liste du
matériel nécessaire, le schéma du montage et les
résultats.
La sécurité dans
l'utilisation du courant électrique occupe une place
prépondérante dans les normes. Dans le domaine
médical, pour assurer la sécurité et
éviter tout risque pour le patient et les opérateurs,
les différentes associations normatives ont publié des
prescriptions précises. La norme IEC 601.1 est le document
général complété ou modifié par un
certain nombre de prescriptions spéciales regroupées
dans les Règles Particulières de Sécurité
des Appareils Electromédicaux
1. LA NORME IEC 601.1
Elle regroupe tous les
types d'appareils médicaux sans distinction, qu'ils soient
électriques ou non. Elle est présentée en dix
sections
- Généralités
: Elle détermine le domaine d'application et l'objet de la
norme. Elle pose la terminologie et les définitions, ainsi
que les caractéristiques générales des
appareils
- Condition
environnement : Elle détermine les
catégories, les moyens, et les conditions de
sécurité
- Protection contre
les chocs électriques : Elle définit la
classification et la protection contre les chocs
électriques, les courants de fuite se rapportant aux
appareils électromédicaux
- Protection contre
les chocs mécaniques : Cette partie traite de la
résistance mécanique, des aspects physiques et
sonores
- Protection contre
les risques dus aux rayonnements non désirables ou
excessifs : Rayonnements X, alpha, bêta, gamma,
micro-ondes, visibles, infrarouges, ultraviolets, énergie
acoustique, et la compatibilité
électromagnétique.
- Protection contre
les risques d'ignition de mélanges anesthésiques
inflammables : Définition de catégories
d'appareils : AP, APG. Essai des appareils.
- Protection contre
les températures excessives et autres risques :
Classification en fonction des risques. Matériaux
utilisés.
- Caractéristiques
: Précision sur les caractéristiques de
fonctionnement et protection contre les caractéristiques de
sortie présentant des risques.
- Fonctionnement
: Conditions de défauts. Essai
d'environnement.
- Règles de
construction : Concernant les constructeurs
2. LA SÉCURITÉ
ÉLECTRIQUE
Avant tout, la norme
précise le domaine d'application, elle pose la terminologie,
ainsi que les caractéristiques générales des
appareils.
A) TERMINOLOGIE ET DÉFINITIONS.
1. Définitions des courants de fuite et auxiliaires
patient.
1.1 Les courants de fuite.
Courants non fonctionnels
traversant ou contournant une isolation. Les courants de fuite
suivants sont définis : courant de fuite à la terre,
courant de fuite à travers l'enveloppe et courant de fuite
patient.
a) Courant de fuite
à la terre :
Courant s'écoulant de
la partie reliée au réseau, en traversant ou
contournant l'isolation, par le conducteur de protection.
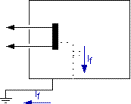
b) Courant de fuite
à travers l'enveloppe :
Courant s'écoulant de
l'enveloppe ou des parties accessibles en utilisation normale
à l'opérateur ou au patient, non comprises les parties
appliquées, par une liaison conductrice extérieure
autre que le conducteur de protection vers la terre ou une autre
partie de l'enveloppe.
c) Courant de fuite
patient :
Courant s'écoulant de
la partie appliquée vers la terre à travers le patient
ou courant s'écoulant du patient vers la terre par une partie
appliquée de type F, dû à la présence non
voulue sur le patient d'une tension provenant d'une source
externe.
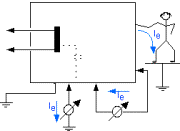
1.2 Les courants auxiliaires patient
Courants s'écoulant
dans le patient en utilisation normale entre des
éléments de la partie appliquée et non
destiné à produire un effet physiologique.
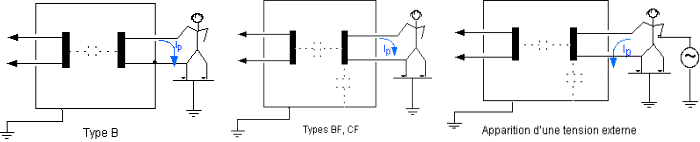
1.3 Tableau des limites.
La norme
générale définit des limites à ces
courants. Les cas particuliers sont définis par les normes
particulières.
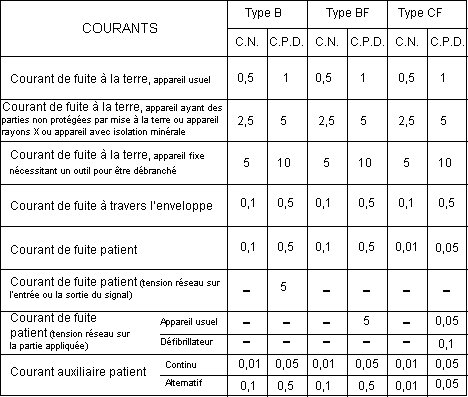
Valeurs exprimées en
milliampères.
C.N. : condition
normale
C.P.D. : condition de premier
défaut
2. Conception de la sécurité
électrique.
Elle a pour but d'isoler le
patient de la terre. La norme définit des classes de
fonctionnement et dans ces classes, des types de
fonctionnement.
2.1 Classes.
a) Appareils de classe
1.
La sécurité
est obtenue par l'isolation et connexion à la terre des
parties métalliques de l'appareil pouvant être sous
tension en cas de défaut.
b) Appareils de classe
2.
La sécurité
est obtenue par une double isolation. Aucune partie métallique
n'est reliée à la terre.
c) Les appareils
alimentés en T.B.T.S. (Très Basse Tension de
Sécurité).
L'alimentation doit
être inférieure à 24 volts en courant alternatif
et 50 volts en courant continu. Il faut distinguer les appareils
à alimentation électrique interne et les appareils
alimentés par l'intermédiaire d'un transformateur
séparé qui pourront être de classe 1 ou de classe
2.
2.2 Les types.
a) Les appareils de type
B.
Tout appareil en contact
avec le patient, excepté les appareils à applications
cardiaques directes. Les circuits en contact avec le patient peuvent
être reliés à la terre.
b) Les appareils de type
BF.
Tout appareil en contact
avec le patient, excepté les appareils à applications
cardiaques directes. Les circuits en contact avec le patient sont de
type Fâ (flottant).
c) Les appareils de type
CF.

Appareil approprié
aux applications cardiaques directes. Les circuits en contacts avec
le patient sont de type F â. Le courant de fuite
toléré est beaucoup plus faible que dans le type
BF.
d) Symboles des
appareils BF et CF protégés contre les effets des
défibrillateurs.

B)
Essais prescrits par la norme.
1. Les différents tests.
- Test de continuité
de la terre de protection.
- Test de résistance
d'isolement entre secteur et enveloppe.
- Test de résistance
d'isolement entre partie appliquée et terre.
- Test de courant de fuite a
la terre - condition normale.
- Test de courant de fuite a
la terre - condition de premier défaut secteur
ouvert.
- Test de courant de fuite a
l'enveloppe - condition normale.
- Test de courant de fuite a
l'enveloppe - condition de premier défaut secteur
ouvert.
- Test de courant de fuite a
l'enveloppe - condition de premier défaut terre
ouverte.
- Test de courant de fuite au
patient - condition normale.
- Test de courant de fuite au
patient - condition de premier défaut secteur
ouvert.
- Test de courant de fuite au
patient - condition de premier défaut terre
ouverte.
- Test de courant de fuite au
patient - condition de premier défaut secteur sur partie
appliquée.
- Test de courant auxiliaire
patient - condition normale.
- Test de courant auxiliaire
patient - condition de premier défaut secteur
ouvert.
- Test de courant auxiliaire
patient - condition de premier défaut terre
ouverte.
- Test de courant de fuite a
l'enveloppe - appareil alimentation interne.
- Test de courant de fuite au
patient - appareil alimentation interne.
- Test de courant de fuite au
patient - condition de premier défaut secteur sur partie
appliquée - appareil alimentation interne.
- Test de courant auxiliaire
patient - appareil alimentation interne.
- Test de courant de fuite a
l'enveloppe - condition normale système
électromédical.
- Test de courant de fuite a
l'enveloppe - condition de premier défaut terre ouverte -
système électromédical.
Pour effectuer ces tests,
les industriels proposent des appareils capables de réaliser
les mesures automatiquement et de sauvegarder les
résultats.
2. Les conditions de test.
- L'appareil doit
être prêt à fonctionner, c'est-à-dire
entièrement fermé.
- L'appareil doit être
testé dans certaines conditions d'humidité de pression
atmosphérique et de température.
- La tension de test doit
être égale à 110 % de la plus haute tension
réseau assignée (voir les spécifications de
l'appareil à tester). Pour ce faire, il est nécessaire
de disposer d'un transformateur d'isolement élévateur
de tension. Les testeurs de sécurité électrique
présents sur le marché mesurent les courants de fuite
sur des appareils qui peuvent consommer jusqu'à 16
Ampère. La puissance du transformateur doit être de 4
à 5 KVA.
- L'appareil doit être
en fonctionnement pendant le test. La plupart ont un autotest
à la mise sous tension ; le testeur de sécurité
électrique doit donc pouvoir introduire un retard à la
mesure.
- Le courant pour la mesure du
circuit de terre doit être de 10 à 25 A sous une tension
de 6 volts pendant 5 secondes.
- Pour les courants de fuite
et auxiliaire patient, le testeur doit pouvoir mesurer les deux
formes de courants continus et alternatifs.
- Toutes les parties
appliquées doivent être vérifiées y
compris celles constituées d'un matériau isolant. Ces
dernières seront mises en contact le plus intimement possible
avec un système conducteur relié électriquement
au testeur. Pour un appareil ayant plusieurs parties
appliquées de ce type, il faut les différencier pour
pouvoir mesurer les courants auxiliaires.
Retour
sommaire annexes
ANNEXE
2
ANALYSE DES EXIGENCES DE
LA NORME ISO 17 025 PAR LA METHODE PIEM
[9]
Retour
sommaire annexes
ANNEXE
3
DÉFINITION DES
OUTILS DE LA PLANIFICATION DE PROJET ET DÉFINITION DES
MACROTACHES
1. Le Processus de
Déroulement du Projet (PDP) :
L'élaboration d'un
Processus de Déroulement du Projet permet de structurer entre
elles et dans le temps les principales tâches à
réaliser pour mener à bien le projet. C'est une vue
d'ensemble du déroulement du projet reprenant les
macrotâches et leur enchaînement dans le temps. On peut y
fait figurer également les jalons d'avancement du projet qui
sont des étapes clefs pour le suivi d'avancement du
projet.
2. Le questionnaire
d'audit selon la norme ISO 17025 :
L'établissement de
ce questionnaire est le point de départ de la
compréhension du contenu du projet. En effet, réaliser
ce questionnaire d'audit permet de prendre conscience de tous les
aspects de la norme et de toutes ses exigences. Il permet aussi
d'extraire les tâches à mettre en oeuvre pour
répondre aux exigences de la norme.
De plus, ce questionnaire
est un moyen d'évaluer concrètement ce qui a
déjà été mis en œuvre et dont la
bonne application a pu être démontrée lors de
l'état des lieux. Mais il joue un rôle également
prépondérant dans la maîtrise de l'avancement de
la démarche d'accréditation. On pourra en effet
utiliser le questionnaire comme point de référence de
l'avancement du projet, puis l'utiliser comme tableau de bord pour
évaluer les évolutions.
3. Le listing des
tâches :
Cet outil consiste
à lister toutes les tâches contenues dans les
macrotâches présentes dans le PDP. Ces tâches
découlent d'une part des exigences de la norme ISO 17025 mais
également des contraintes organisationnelles et des
compétences nécessaires pour gérer un tel
projet. (voir détail)
4. L'Analyse des
Modes de Défaillances, de leurs Effets et de leur
Criticité (AMDEC) :
Cet outil a pour but
d'extraire les risques majeurs associés à la
réalisation d'un projet. On analyse chaque étape issue
du listing des tâches et on relève les
possibilités de défaillance et leurs effets sur le
déroulement du projet. On associe une criticité qui
dépend de la gravité de la défaillance sur le
déroulement du projet, de sa probabilité d'apparition
et de sa probabilité de non-détection, à chaque
tâche analysée et on extrait des points critiques pour
lesquels on essaie de trouver des solutions.
5. Le tableau
d'identification des pré requis et des formations
:
Le but de cet outil
est de regrouper dans un tableau les pré requis
nécessaires pour réaliser chaque tâche et les
formations possibles pour satisfaire ces pré requis. Dans ce
cadre, chaque formation devient une pré-tâche à
la tâche dont elle a pour but de satisfaire les pré
requis.
6. Le tableau
d'identification et d'affectation des ressources :
Pour réaliser
un projet, il est nécessaire de disposer de ressources
humaines et temporelles. Le but de cet outil est d'associer à
chaque tâche un temps de réalisation et la
répartition de ce temps entre les diverses ressources
disponibles et compétentes pour réaliser la tâche
considérée.
7. Les plannings de
Gantt :
Le planning de Gantt
représente l'enchaînement temporel des tâches. Les
tâches peuvent s'effectuer les unes à la suite des
autres ou simultanément en fonction des ressources disponibles
et de la tâche considérée. On fait figurer sur le
diagramme de Gantt tous les jalons d'avancement qui permettent
d'effectuer des bilans intermédiaires de l'avancement du
projet.
8. Définition
des macrotâches du projet
Définition des
tâches contenues dans chaque macrotâche du PDP. Afin de
pouvoir obtenir l'accréditation ISO 17025.
- T0 : Formation à
la norme
- T1 : Diagnostic du
laboratoire
- T2 :
Planification
- T3 : Mise en
œuvre
- T4 :
Validation
- T5 : Demande
d'audit
L'écriture suivante
a été adoptée : chaque tâche commence par
la lettre T. Elle est rattachée à la macrotâche
à laquelle elle appartient. On hiérarchise ensuite les
tâches entre elles par un numéro secondaire. Cela ne
définit pas forcément un ordre de réalisation
dans le temps. Les jalons d'avancement commencent par la lettre J et
sont également rattachés à leur
macrotâche.
T0 : Formation
à la norme
- T0.0 : Lecture de la
norme
- T0.1 : Transfert de
connaissance avec tous les groupes
- J0.0 : Bilan de la
formation à la norme
T1 : Diagnostic du
laboratoire
- T1.0 : Diagnostic
à l'aide du questionnaire ISO17025
- J1.0 : Evaluation de la
faisabilité du projet
T2 : Planification :
(travail préparatoire de l'équipe
GE37)
- T2.0 : Formation
à la norme
- T2.1 :
Réalisation du questionnaire d'audit
- T2.2 : Formation
sécurité électrique
- T2.3 :
Définition des tâches
- T2.4 : Identification
des méthodes de formations aux
pré-requis
- T2.5 : Priorisation des
tâches
- T2.6 : Analyse des
risques
- T2.7 : Etablir une
procédure de communication
- T2.8 : Fourniture d'un
support didactique
- J2.0 : Revue de
bilan
T3 : Mise en
œuvre
- T3.0 :
Réalisation des tâches
- T3.0.1 : Validation ou
mise en place d'un système qualité
- T3.0.2 : Validation ou
mise en place d'un système de gestion de la
documentation
- T3.0.3 : Gestion de la
sous-traitance des essais et des étalonnages
- T3.0.4 : Validation ou
mise en place des processus de gestion des achats
- T3.0.5 : Mise en place
de processus pour la gestion de la clientèle
- T3.0.6 : Mise en place
d'une procédure de réclamation
- T3.0.7 : Mise en place
de processus visant à maîtriser des travaux non
conformes
- T3.0.8 : Mise en place
de procédures d'actions correctives
- T3.0.9 : Mise en place
de procédures d'actions préventives
- T3.0.10 : Mise en place
de procédure visant à maîtriser les
enregistrements (ex : matériel)
- T3.0.11 : Mise en place
d'un calendrier d'audit interne
- T3.0.12 : Mise en place
d'un calendrier de revues de directions
- T3.0.13 : Mise en place
de la gestion du personnel
- T3.0.14 :
Vérification des installations et des conditions
ambiantes
- T3.0.15 : Mise en place
et validation des méthodes de test
- T3.0.16 :
Vérification de l'équipement
- T3.0.17 :
Rédaction des procédures :
-de manutention,
-d'assurance de la
qualité
- T3.0.18 : Mise en place
de la gestion des rapports sur les résultats
- J3.0.0 : Bilan de la
réalisation
T4 : Validation
:
- T4.0 : Audit à
Blanc
- T4.1 :
Correction
- J4.0 : Bilan des
actions correctives
T5 : Demande
d'audit
- T5.0 : Contacter la
personne concernée
Les tâches
constituant T3 sont des tâches issues directement des exigences
contenues dans la norme ISO 17025.
Retour
sommaire annexes
ANNEXE
4
LISTE DES PROCEDURES A
ETABLIR POUR REPONDRE AUX EXIGENCES.
4- prescription
relatives au management.
4.1-
organisation.
4.1.5- procédures pour la protection des informations
confidentielles et des droit de propriétés des
clients.
Procédures protégeant la transmission et le stockage
électronique des résultats.
4.3- maîtrise de
la documentation.
4.3.1- procédures de maîtrise de tous les
documents.
4.3.2.1- procédure identifiant le statut de révision en
cours et la diffusion des documents.
4.3.3.4- procédures décrivant comment les modifications
dans les documents conservés dans les systèmes
informatiques sont effectuées et
maîtrisées.
4.4- revue des demandes.
appel d'offres et contrats.
4.4.1- procédures pour la revue des demandes. des appels
d'offre. des contrats.
4.6- achat de services
et de fournitures.
4.6.1- procédures pour la sélection et l'achat des
services et des fournitures utilisés et qui ont une incidence
sur la qualité des essais et/ou étalonnages.
4.8-
réclamations
procédure pour traiter les réclamations clients ou
autres parties.
4.9- maîtrise des
travaux non conformes
4.9.1- procédures de non-conformité des travaux
d'essais et/ou d'étalonnage.
4.10- actions
correctives.
4.10.1- procédure pour la mise en œuvre d'actions
correctives des travaux non conformes ou des
écarts.
4.11- actions
préventives.
4.11.2- procédures relatives aux actions
préventives.
4.12- maîtrise des
enregistrements.
4.12.1- généralités.
4.12.1.1- procédures d'identification de collectes.
d'indexages. d'accès. de classement. de stockage. de
conservation. d'élimination des enregistrements techniques et
relatifs à la qualité.
4.12.1.4- procédures de protection et sauvegarde des
enregistrements stockés électroniquement.
4.14- revue de
direction.
4.14.1- procédure de revue du système qualité et
des activités d'essai et/ou d'étalonnage.
5- prescriptions
techniques
5.4- méthode
d'essai et d'étalonnage et validation des
méthodes.
5.4.1- procédures appropriées pour tous les essais
et/ou étalonnages.
5.4.6- estimation de l'incertitude des mesures de tous les
étalonnages.
5.4.6.1- procédure d'estimation de l'incertitude de mesure de
tous les étalonnages.
5.4.7- maîtrise des données.
5.4.7.2- procédures de protection des
données.
5.5-
équipements.
5.5.6- procédures pour la manutention sûre. le
transport. le stockage. l'utilisation et la maintenance
planifiée des instruments de mesure.
5.5.11- procédures pour assurer que les copies sont
correctement mises à jour.
5.6-
traçabilité du mesurage.
Procédure d'étalonnage de l'équipement du
laboratoire d'essai et/ou d'étalonnage.
5.6.3- étalons de référence et matériaux
de référence
5.6.3.1- procédure pour l'étalonnage des étalons
de référence.
5.6.4.3- procédures de vérifications
intermédiaires d'étalonnage.
5.6.3.4- procédures pour la manutention. le transport. le
stockage et l'utilisation des étalons de
références.
5.7-
échantillonnage.
5.7.1- procédures documentée
d'échantillonnage.
5.7.3- procédures d'enregistrement des données
pertinentes et des opérations se rapportant à
l'échantillonnage.
5.8- manutention des
objets d'essai et d'étalonnage.
5.8.1- procédures pour le transport. la réception. la
protection. le stockage. la conservation et/ou l'élimination
d'objet d'essai et/ou d'étalonnage.
5.8.4- procédures pour éviter la
détérioration. la perte ou l'endommagement de l'objet
d'essai et d'étalonnage lors du stockage. de la manutention et
de la préparation.
5.9- assurer la
qualité des résultats d'essai et
d'étalonnage.
procédures de
maîtrise de la qualité pour surveiller la
validité des essais et étalonnages
entrepris.
Retour
sommaire annexes
ANNEXE
5
COMPARATIF ISO 9001 ET ISO
17025
Il existe de nombreuses
corrélations entre le référentiel ISO 9001 et le
référentiel ISO 17025 :
|
ISO 9001 : 2000
|
ISO
17025
|
|
5.1 / 5.3 /
5.4.1
|
4.1.3 /
4.2.2
|
|
5.5.1
|
4.1.5 / 4.2.4
/ 4.9.1 / 4.10.1 / 5.2.5
|
|
6.1 /
6.2.1
|
4.1.5 /
5.5.1
|
|
5.5.2
|
4.1.5
|
|
5.6.1 /
8.5.1
|
4.14
|
|
4.1 / 4.2.1 /
4.2.2
|
4.2.1 / 4.2.2
/ 4.2.3
|
|
5.4.2 /
7.1
|
4.2.1 / 4.2.2
/ 4.14
|
|
5.2 /
7.2
|
4.4
|
|
7.3
|
1.5 / 5.4.2 /
5.5.3 / 5.4.4 / 5.4.5
|
|
4.2.3
|
4.3 / 5.4.7 /
5.5.11
|
|
7.4
|
4.6 / 5.5 /
5.6.1 / 5.6.2.1 / 5.6.2.2
|
|
7.4.1
|
4.5 /
4.6
|
|
7.4.2
|
4.6
|
|
7.4.3
|
4.5 / 4.6.4 /
4.7 / 5.5.2
|
|
7.5.4
|
5.8 /
5.10.6
|
|
7.5.3
|
5.5.4 /
5.8
|
|
6.3 / 6.4 /
7.5.1 / 7.5.2
|
4.12 / 5.3 /
5.5 / 5.8 / 5.9
|
|
7.1 /
8.1
|
5.4
|
|
7.4.3 /
8.2.4
|
4.5 / 4.6 /
5.5.2 / 5.8
|
|
8.2.4
|
4.9 / 5.5.9 /
5.8.3 / 5.8.4 / 5.9
|
|
8.2.4
|
5.4.7 / 5.9 /
5.10.1
|
|
7.5.3 /
8.2.4
|
4.12.2
|
|
7.6
|
5.4 / 5.5 /
5.6
|
|
7.6
|
5.3 / 5.4.1 /
5.4.5 / 5.5 / 5.6
|
|
7.5.3
|
5.5.12 / 5.8
/ 5.9.2
|
|
8.3
|
4.9
|
|
8.5
|
4.10 /
4.11
|
|
7.5.5 /
7.5.1
|
5.9
|
|
4.2.4
|
4.12
|
|
8.2.2 /
8.2.3
|
4.10.5 /
4.13
|
|
6.2.2
|
5.2 /
5.5.3
|
|
7.5.1
|
4.7 / 5.2.1 /
5.10.5
|
|
8.1 / 8.2.3 /
8.2.4 / 8.4
|
5.9
|
Retour
sommaire annexes
 2.
PRESCRIPTIONS RELATIVES AU MANAGEMENT.
2.
PRESCRIPTIONS RELATIVES AU MANAGEMENT.