|
Avertissement
|
|
Si vous arrivez directement sur cette
page sachez que ce travail est un rapport
d'étudiant(e)s et doit être pris comme tel. Il
peut donc comporter des imperfections ou des
imprécisions que le lecteur doit admettre et donc
supporter. Il a été réalisé lors
du semestre d'enseignement théorique à
l'UTC
et constitue avant-tout un travail de
compilation
bibliographique, d'initiation et d'analyses sur des
thématiques associées aux technologies
biomédicales.
Nous ne faisons aucun
usage commercial et la duplication est libre. Si vous avez
des raisons de contester ce droit d'usage, merci
de nous en faire
part.
L'objectif de la présentation sur le Web est de
permettre l'accès à l'information et
d'augmenter ainsi les échanges professionnels. En cas
d'usage du document, n'oubliez pas de le citer comme source
bibliographique. Bonne lecture...
|
|
Réference
à rappeler :
Marquage CE des dispositifs
médicaux, I. Caro, R. Lebret, Projet DESS "TBH", UTC,
1997, pp 30,
URL :
https://www.utc.fr/~farges/DESS_TBH/96-97/Projets/CE/Marqu_CE.htm
|
MARQUAGE
CE
DES DISPOSITIFS MEDICAUX
(COMPARAISON DES ORGANISMES
NOTIFIES EN FRANCE ET AU ROYAUME-UNI)
|

|

|
|
Isabelle
CARO
|
Richard
LEBRET
|
|

1) Introduction
2) Le marquage CE
2.1. Historique
des Directives
2.2. Champ
d'application des Directives
2.3. Répartition
des rôles
2.4. Détermination
de la classe du dispositif
2.5. Les
procédures d'évaluation de la conformité au
marquage CE
3) Le système
britannique
3.1. Le MDA (Medical Devices
Agency)
3.2. Les "Notified Bodies" (organismes
notifiés )
4) Le
système français
4.1. La structure
du G-Med
4.2. Participation
aux réglementations
5) Comparaison
des systèmes
6) Le comportement des fabricants
7) Conclusion
générale
Glossaire
Bibliographie Annuaire Questionnaire
Les nouvelles directives 90/385/CE et 93/42/CE rendent obligatoire
le marquage CE des dispositifs médicaux pour leur libre
circulation dans l'Espace Economique Européen (EEE). Ces
nouvelles directives ont impliqué la mise en place de nouveaux
acteurs : les organismes notifiés . Ces
derniers effectuent la validation technique des dispositifs
médicaux pour l'apposition du marquage CE. La France et le
Royaume- Uni ont une politique différente en
ce qui concerne le nombre de leurs organismes notifiés. En
effet, la France ne s'est dotée que d'un seul
organisme notifié alors que le Royaume-uni en a
plusieurs (neuf jusqu'à présent).
Après une introduction sur le marquage CE, nous traiterons de
l'objet de ce projet, qui est de présenter et de
confronter les systèmes français et anglais
pour l'application du marquage CE des dispositifs médicaux.
Nous nous intéresserons aux comportements des fabricants
anglais et français de dispositifs médicaux, face au
choix d'un organisme notifié sachant qu'ils
sont libres de faire effectuer le contrôle de leurs dispositifs
par n'importe quel organisme notifié européen. Nous
avons pour cela, établi un
questionnaire que nous avons envoyé a des
entreprises implantées en France et au Royaume-Uni, et nous
présenterons l'analyse de leurs réponses.
(Retour
sommaire)
2.1. Historique des Directives :
"L'Acte Unique" signé en décembre
1985 engage les Etats Membres de la Communauté Economique
Européenne à créer un espace sans
frontières intérieures pour permettre la libre
circulation des personnes, des biens, des capitaux et des
services.
Au niveau des dispositifs médicaux, les entraves techniques
qui existaient d'un pays à l'autre ne pouvaient être
supprimées qu'en substituant aux réglementations
nationales une règlementation communautaire
harmonisée.
C'est ainsi qu'une "Nouvelle Approche" a été
définie en vue de la réalisation du "Marché
Unique" et c'est dans cet esprit que trois directives ont
été prévues pour l'ensemble des dispositifs
médicaux. Elles sont respectivement relatives aux :
- dispositifs médicaux implantables
actifs (directive 90/385/CEE).
Obligatoire depuis le 1er Janvier 1995.
- autres dispositifs médicaux (directive
93/42/CEE).
Applicable depuis le 1er Janvier 1995 et obligatoire à
partir du 14 Juin 1998.
- dispositifs destinés au diagnostic
in-vitro (en cours d'élaboration).
Ces différentes Directives aboutissent à
l'obligation du Marquage CE pour la mise sur le marché des
dispositifs médicaux dans l'Espace Economique Européen
(EEE). Celui-ci comprend les pays membres de l'Union
Européenne et les pays membres de l'Association
Européenne de Libre Echange (AELE). Le marquage CE permet
alors à un dispositif médical de circuler librement
dans l'EEE. Il est renouvelable
tous les cinq ans.
Transposition en droit national :
Les Etats Membres doivent intégrer les dispositions
contenues dans les directives à leurs législations et
réglementations. En France, la loi n 94/43 du 18 Janvier 1994
modifiée par la loi du 4 Février 1995 transpose les
directives 90/385/CEE et 93/42/CEE en droit
français.
(Retour
sommaire)
2.2. Champ d'application des Directives :
La Directive 93/42/CEE s'applique aux dispositifs
médicaux et à leurs accessoires qui répondent
aux définitions suivantes :
Dispositif médical : Tout instrument,
appareil, équipement, matière ou autre article,
utilisé seul ou en association, y compris le logiciel
nécessaire pour le bon fonctionnement de celui-ci,
destiné par le fabricant à être utilisé
chez l'homme à des fins :
- de diagnostic, de prévention, de
contrôle, de traîtement ou d'atténuation
d'une maladie.
- de diagnostic, de contrôle, de traitement,
d'atténuation ou de compensation d'une blessure ou d'un
handicap.
- d'étude ou de remplacement ou modification de
l'anatomie ou d'un processus physiologique.
- de maîtrise de conception.
et dont l'action principale voulue dans ou sur le corps humain
n'est pas obtenue par des moyens pharmacologiques ou
immunologiques ni par métabolisme, mais dont la fonction
peut être assistée par de tels moyens.
Accessoire : Tout article qui est destiné
principalement par son fabricant à être
utilisé avec un dispositif médical afin de permettre
l'utilisation de ce dispositif, conformément aux intentions
de son fabricant.
La Directive 90/385/CEE s'applique aux dispositifs médicaux
implantables actifs (D.M.I.A.) définis ainsi :
Dispositifs médicaux implantables actifs :
Tout dispositif médical dépendant pour son
fonctionnement d'une source d'énergie électrique ou
de toute autre source d'énergie que celle
générée directement par le corps humain ou la
pesanteur, qui est conçu pour être implanté en
totalité ou en partie, par une intervention chirurgicale ou
médicale, dans le corps humain ou, par une intervention
médicale, dans un orifice naturel et qui est destiné
à rester après l'intervention.
(Retour sommaire)
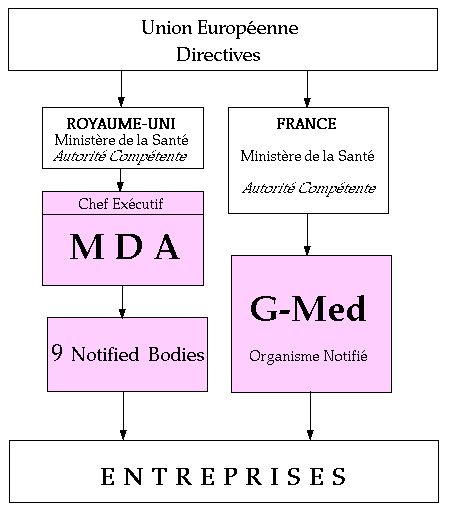
2.3. Répartition des
rôles
Dans chaque pays de l'EEE, l'Autorité Compétente (le
Ministère de la Santé) est chargée de mettre en
place les Directives.
L'Autorité Compétente doit donc :
- désigner les organismes notifiés.
- assurer la mise en place du système de
matério-vigilance.
- intervenir en cas de litige entre un fabricant et un organisme
notifié.
Les Directives définissent des procédures de
conformité à des Exigences Essentielles
auxquelles doivent satisfaire les dispositifs. Ces Exigences
Essentielles sont définies dans l'annexe I de la directive
93/42/CEE et elles portent sur :
- les conditions d'utilisation clinique et de
sécurité pour le patient et les utilisateurs.
- l'évaluation et l'acceptibilité des risques.
- la fonctionnalité de dispositif.
- la pérennité du dispositif.
- les conditions de protection durant le stockage et/ou le
transport.
Pour certaines classes de dispositifs, les procédures de
conformité prévoient le recours à une tierce
partie indépendante appelée Organisme Notifié .
Les certificats alors délivrés par cette tierce partie
permettent l'apposition du Marquage CE et, par la même, la mise
sur le marché dans l'EEE des dispositifs concernés.
Les organismes notifiés, désignés par
l'Autorité Compétente de leur pays, sont
accrédités pour effectuer une ou plusieurs
procédures de conformité aux exigences essentielles
décrites dans les annexes des Directives. Ils doivent
répondre aux normes des procédures qu'ils
préconisent (série EN45000).
Les organismes notifiés doivent :
- être indépendants
- être impartiaux
- être responsables
- avoir les équipements adéquates
- faire voeux de confidentialité
Enfin, chaque organisme notifié possède un
numéro d'identification de trois chiffres que l'on
retrouvera sur les dispositifs qu'il certifie. Le marquage CE
consiste donc en deux lettres CE + trois chiffres (ex: CE 459 pour le
G-Med).
Un fabricant peut s'adresser à n'importe quel organisme
notifié européen. Le fabricant répond
également à une définition très
précise :
C'est la personne physique ou morale responsable de la
conception, de la fabrication, du conditionnement et de
l'étiquetage d'un dispositif médical en vue de sa
mise sur le marché en son propre nom.
Parallèlement aux directives, les Exigences Essentielles
sont traduites en spécifications techniques par des normes
européennes harmonisées. Ainsi, un dispositif
respectant ces normes (non obligatoires) est présumé
respecter les Exigences Essentielles et il peut donc être
marqué CE. A ce jour, la plupart des procédures de
conformité aux exigences essentielles possède une norme
qui la reprend (ex: l'annexe II pour le système complet
d'assurance qualité est traduite par la Norme NF EN
46001).
(Retour sommaire)
2.4. Détermination de la classe du
dispositif
Les dispositifs qui sont du ressort de la directive 93/42 sont
répartis en quatre classes : I, IIa, IIb et III,
suivant le niveau de risque du dispositif. La classification se fait
conformément aux règles figurant dans l'annexe IX de
cette Directive.
Ces règles s'appuient sur un certain nombre de critères
:
- la durée
d'utilisation ,
- les caractères invasifs ou non , et type
d'invasivité,
- la possibilité ou non de ré-utilisation,
- la visée thérapeutique ou
diagnostique ,
- la dépendance ou non de la source
d'énergie ,
- la partie du corps en contact avec le dispositif.
Ces règles, qui sont au nombre de 18, peuvent être
réparties en cinq grands groupes :
- Règles 1 à 4 :
dispositifs non invasifs.
- Règle 5 : dispositifs invasifs par un
orifice du corps.
- Règles 6 à 8 : dispositifs
invasifs par voie chirurgicale.
- Règles 9 à 12 : règles
additionnelles sur les dispositifs actifs.
- Règles 13 à 18 : règles
spéciales visant les catégories particulières
de dispositifs
L'ensemble de ces règles permet de déterminer
la classe d'un dispositif médical.
Il est important de signaler que ce classement est effectué
par le fabricant. Cependant, la détermination de la classe
d'un dispositif étant parfois difficile, les organismes
notifiés assistent souvent les fabricants pour classer les
dispositifs.(Retour sommaire)
Remarques importantes pour la classification d'un dispositif
:
- Si le dispositif est destiné à être
utilisé en combinaison avec un autre dispositif, les
règles de classification s'appliquent
séparément à chacun des dispositifs. Les
accessoires sont classés en tant que tels,
indépendamment de dispositifs
médicaux avec lesquels ils sont utilisés.
- Le logiciel informatique commandant un dispositif ou agissant
sur son utilisation relève automatiquement de la même
classe.
- Si le dispositif n'est pas destiné à être
utilisé exclusivement ou essentiellement dans une partie
spécifique du corps, il doit être
considéré et classé suivant l'utilisation la
plus critique telle que
spécifiée.
- Si plusieurs règles s'appliquent au même dispositif
du fait des utilisations indiquées par le fabricant, la
règle qui s'applique est la plus stricte ,
le dispositif étant dans la classe la plus
élevée.
(Retour
sommaire)
2.5. Les procédures d'évaluation
de la conformité au marquage CE
Le mode de preuve de la conformité des appareils
médicaux au marquage CE est fonction de la classe dans
laquelle ces derniers se rangent :
- Si le dispositif est de classe I , la
procédure est une "auto-certification" du fabricant ne
faisant pas appel à un organisme notifié. Les
dispositifs médicaux de cette classe ne requierent
pas l'intervention d'un organisme notifié , sauf
dans le cas où le dispositif est mis sur le marché
à l'état stérile, ou qu'il comporte une ou
plusieurs fonctions de mesurage.
Ci-dessous, mode de preuve : classe I
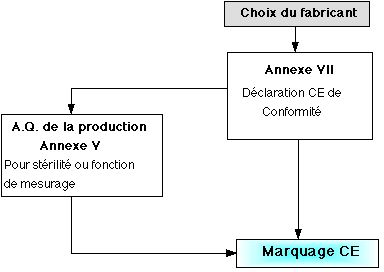
(Retour sommaire)
- Si le dispositif est de classe IIa, IIb ou
III , le fabricant devra choisir une procédure
d'évaluation du dispositif faisant appel à un
organisme notifié.
Ci-dessous, mode de preuve : classe IIa
(Intervention obligatoire d'un organisme notifié)
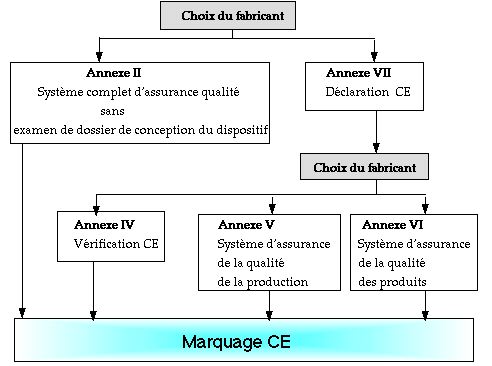 (Retour
sommaire)
(Retour
sommaire)
Ci-dessous, mode de preuve : classe IIb
(Intervention obligatoire d'un organisme notifié)

(Retour sommaire)
Ci-dessous, modes de preuve : classe III et D.M.I.A.
(Intervention obligatoire d'un organisme notifié)
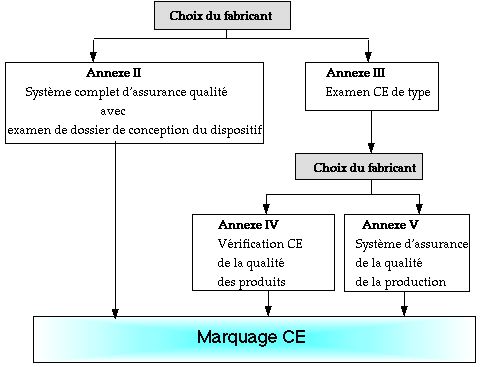
Annexes reprises par des normes :
Annexe II : NORME NF EN 46001
Annexe V : NORME NF EN 46002
Annexe VI: NORME ISO 9003
Annexe VII : Documentation technique :
- Description du produit,
- dessin et schémas de conception,
- listes des normes utilisées,
- rapport d'essais,
- données cliniques.
(Retour
sommaire)
|
3. Le système britannique
|
3.1. Le "Medical
Devices Agency" (MDA)
Dans chaque pays de l'Espace Economique Européen,
l'Autorité Compétente agit au nom du gouvernement pour
s'assurer de la bonne exécution des directives relatives aux
dispositifs médicaux.
Au Royaume-Uni, l'Autorité Compétente, le
Ministère de la Santé, a délégué
sa responsabilité au MDA. Le MDA a été
créé le 27 Juillet 1994. C'est une agence
exécutive du Département de la Santé britannique
qui est chargée de protéger la Santé
Publique et sauvegarder les intérêts des patients et des
utilisateurs .
L'agence est dirigée par un Chef Exécutif nommé
par le Ministre de la Santé britannique à qui il doit
rendre compte du bon fonctionnement de l'agence.
Outre son statut d'Autorité Compétente pour les
"Affaires Européennes" , le MDA a d'autres
services :
" Device Technology & Safety (DTS) "
: Cette cellule est responsable de la
Matério-vigilance. De plus, le DTS fournit des conseils
techniques sur les dispositifs afin de prévenir les
accidents.
" Manufacturer Registration Scheme (MRS) " :
Cette cellule audite les systèmes d'assurance
qualité des fabricants de dispositifs médicaux
vendus aux hôpitaux. Elle publie également un
registre des compagnies en règle. Le MRS est en voie de
disparition avec l'apparition des nouvelles directives.
" Medical & Nursing (MN) " : Cette cellule
réalise des expertises diverses pour toutes les autres
cellules de l'agence.
(Retour sommaire)
170 personnes travaillent au MDA. On y trouve des
spécialistes en médecine, en biologie, en chimie, en
physique, en pharmacie ainsi que des spécialistes en assurance
qualité. La répartition du temps de travail du
personnel du MDA est la suivante :
13 % : Affaires Européennes (directives, marquage
CE...)
7 % : Etablissements des normes
16 % : Manufacturer Registration Scheme (MRS)
27 % : Matério-vigilance
16 % : Conseils
9 % : Evaluation des dispositifs
12 % : Support des différentes activités
Le MDA définit le rôle de l'Autorité
Compétente ainsi :
Sauvegarder la Santé Publique en assurant que les
dispositifs médicaux vendus et utilisés dans son
pays rencontrent les critères de sécurité, de
qualité et de performance appropriés.
En temps qu'Autorité Compétente pour le Royaume-Uni,
le MDA a 5 fonctions principales :
* Faire respecter les nouvelles règlementations
relatives à la mise en pratique des directives.
* Fournir des conseils aux fabricants et aux
utilisateurs sur les directives.
* Diriger le "Système de Vigilance" en cas d'incident
survenant sur un dispositif (Matério-vigilance).
* Evaluer les protocoles d'investigations cliniques de nouveaux
dispositifs.
* Nommer et surveiller les Organismes Notifiés
(Notified Body en anglais).
(Retour sommaire)
Avant de nommer un "Notified Body" , le MDA :
* Inspectera l'organisme en question en s'assurant de sa
bonne connaissance des dispositifs médicaux et supervisera
l'audit d'un système d'assurance qualité
réalisé par cet organisme.
* Conseillera les procédures d'audit des organismes
notifiés.
* S'assurera que les directives soient bien reprises par les
procédures d'audit qui devront être ni trop
sévères, ni trop laxistes.
Après avoir accrédité un "Notified
Body" , le MDA a un droit de regard sur ses
activités grâce à des audits annuelles qui
peuvent, le cas échéant, remettre en cause le statut de
"Notified Body".
Le MDA ayant l'expérience d'audits grâce au
système anglais d'homologation (MRS : Manufacturer
Registration System), il a le souci de former les différents
"Notified Bodies" à l'audit des fabricants de
dispositifs médicaux. Il organise des séminaires
à cet effet.
En cas de litige entre un fabricant et un "Notified Body", le MDA a
un rôle de conseiller. Pour les dispositifs ne faisant
pas appel à un organisme notifié (classe I), le MDA est
tenu d'enregistrer ces "autocertifications". Enfin, le MDA
négocie actuellement l'établissement de la
troisième directive relative au diagnostic in-vitro avec les
autres membres de l'EEE.
(Retour sommaire)
3.2. Les "Notified Bodies "(organismes
notifiés anglais)
Les "Notified Bodies" sont désignés par le
MDA selon les critères de la série des normes EN 45000
et le code de conduite de celui-ci décrit
précédemment.
Chaque "Notified Body" est qualifié pour effectuer les
procédures des annexes pour lesquelles il est
accrédité. Les contrôles d'un "Notified Body"
peuvent donc être limités à un certain type de
dispositifs selon leurs compétences.
Jusqu'à présent, le MDA a désigné
9 "Notified Bodies" pour le Royaume-Uni. Voici un
tableau synthétique des annexes et des dispositifs couverts
par les différents "Notified Bodies" :
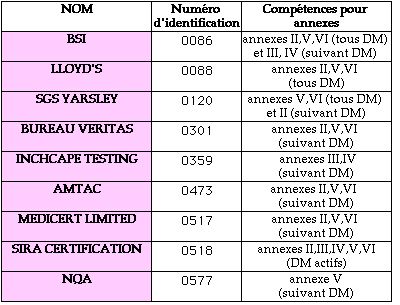
DM : Dispositifs Médicaux
(Retour sommaire)
Cette pluralité implique une concurrence entre tous les
organismes notifiés. Chaque organisme a donc une politique
commerciale développée. La rapidité avec
laquelle le MDA et les "Notified Bodies" nous ont répondu
témoigne d'une "aggressivité"
commerciale que nous n'avons pas ressentie en contactant le
G-Med.
Notre enquête sur ces différents organismes nous a
permis de recueillir quelques informations sur certains d'entre eux
:
AMTAC : Ce fut le premier à
être accrédité (par le NAMAS : service
d'accréditation). Il réalise la certification des
systèmes Qualité ISO 9000, EN 29000 et EN
46001/2.
60 personnes travaillent dans le département "dispositifs
médicaux". Il propose d'inclure la certification ISO 9000
avec le marquage CE. Enfin, il prend en compte les certifications
préalables (MRS,BSI) pour le marquage CE.
BSI : C'est le plus important des Notified
Bodies. (Il est accrédité pour toutes les annexes).
Il réalise également les certifications d'assurance
Qualité ISO 9000.
NQA : Il n'est accrédité que pour
les systèmes d'assurance qualité. Les dispositifs
médicaux ne concernent qu'un pourcent de leurs
activités.
SIRA : Il ne contrôle que les dispositifs
médicaux actifs.
Remarque : la totalité des adresses des "notified
bodies" et l'adresse du MDA sont consultables dans l'annuaire
en fin de rapport.
(Retour sommaire)
L'Autorité Compétente française est le
Ministère de la Santé. A ce titre, il a
désigné un organisme notifié, le G-Med, pour
faire appliquer les directives 90/385/CEE et 93/42/CEE. Le G-Med a la
clé du marché pour tous les dispositifs
médicaux. Le G-Med est le seul organisme
notifié français . Son numéro
d'identification en tant qu' organisme notifié est le
0459.
4.1. La structure du G-Med
Le G-Med a été créé le 1er Mars 1994 sous
la forme d'un groupement d'intérêt économique
entre le Laboratoire Central des Industries Electriques (LCIE), le
laboratoire national d'essais (LNE) et les Ministères de
l'Industrie et de la Santé.
Le LCIE et le LNE étaient déjà associés
depuis plus de 15 ans au sein du GLEM (Groupement des Laboratoires
d'Essais des Matériels de techniques médicales) dans le
cadre de l'homologation.
Le GLEM avait comme fonctions :
* L'assistance technique des industriels,
* La certification volontaire des industriels.
Le système d'évaluation des dispositifs
médicaux imposées par les nouvelles directives ont
modifié les activités du G-Med. En effet, leurs
activités techniques ont diminué pour laisser place
à des activités d'audits et de conseils
d'entreprises . Ce changement d'activité est la cause
de la diminution de leur effectif, qui est passé de 60
à 37 personnes depuis l'application des
directives.
La participation des Ministères de l'Industrie et de la
Santé permet une bonne communication entre les
Ministères et l'organisme notifié. Le G-Med en tant
qu'organisme notifié est contrôlé par le
COFRAC.
L'activité du G-Med se repartit en 3 grandes zones
géographiques:
- 40 % en France,
- 20 % en Europe,
- 40 % dans le reste du monde.
(Retour sommaire)
Le G-Med a des accords avec l'AFAQ
(Association Française d'Assurance Qualité). En effet,
il tient compte de ses rapports d'audits effectués au titre de
la certification volontaire des systèmes de qualité
(ex: ISO 9001). Cette prise en compte signifie que les exigences
essentielles sont présumées satisfaites et ne sont pas
vérifiées au cours des audits complémentaires
destinés à évaluer la conformité aux
autres exigences. La politique actuelle du G-Med tend à
développer ce genre de collaboration avec d'autres organismes.
Ce système de prise en compte étant attractif pour le
fabricant.
Le G-Med appose également la norme NF (norme
française) sur les appareils médicaux. On peut
différencier ces deux activités en disant que, le
marquage CE évalue en premier lieu la sécurité
alors que la norme NF évalue la fonctionnalité d'un
appareil.
4.2. Participation aux réglementations
:
Le G-Med participe activement aux travaux nationaux,
européens, et internationaux de normalisation. Ses relations
privilégiées avec les autorités
réglementaires et son réseau de partenaires
français et étrangers, lui permettent d'évaluer
les exigences essentielles de sécurité ainsi que les
performances. Il tient compte des derniers développements de
techniques et de la normalisation.
Par ailleurs, en dehors des prestations strictement
nécessaires au Marquage CE, le G-Med
offre ses services pour:
* la certification des systèmes de qualité
selon les référentiels normatifs de la série
NF EN 46000,
* le pré-audit des systèmes d'assurance
qualité en vue de préparer ceux-ci à une
certification ultérieure,
* les essais et examens techniques, à la demande du
fabricant, en vue de vérifier la conformité à
des normes,
* des examens ou essais techniques en vue de délivrance du
droit d'usage des marques volontaires (NF Médicale),
* une formation des personnels industriels (fabricants,
distributeurs,...) ou utilisateurs, aux conséquences des
directives et de leurs exigences,
* l'audit des système d'assurance de la qualité des
sous-traitants, en particulier des sous-traitants de
stérilisation.
(Retour
sommaire)
|
5. Comparaison des deux
systèmes
|
L'Autorité Compétente de chaque pays de l'Espace
Economique Européen (EEE) doit mettre en place l'application
des directives. Ainsi, pour effectuer les procédures de
conformité aux Exigences Essentielles, la France a choisi de
ne désigner qu'un seul organisme notifié alors que le
Royaume-Uni a fait le choix d'en désigner plusieurs.
L'organigramme comparatif des systèmes français et
anglais:
Le choix français :
La France a choisi de faire confiance au G-Med qui peut s'appuyer
sur son expérience acquise par le système
français d'homologation qu'il continuera de contrôler
jusqu'à sa disparition en juin 1998. Sa compétence pour
l'ensemble des dispositifs médicaux et l'ensemble des annexes
n'incitait pas l'Autorité Compétente à susciter
d'autres organismes.
Le choix britannique :
Au Royaume-Uni, le MDA jouit de la même expérience
avec leur système interne d'homologation volontaire (MRS)
qu'il continue d'effectuer jusqu'en juin 1998 également.
Toutefois, leur statut d'Autorité Compétente
hérité du Ministère de la Santé les a
amené à désigner un certain nombre d'organismes
notifiés pour appliquer les procédures de
conformité. Les organismes qui ont postulé au nouveau
statut de "Notified Body" sont pour la plupart de grands Bureaux
Certificateurs qui ont une expérience des audits de
systèmes d'assurance qualité ISO (BSI, Veritas,
Lloyd's...).
La comparaison :
La première différence qui ressort de la comparaison
est le passé des organismes : Le G-Med a toujours
baigné dans le milieu médical alors que les "Notified
Bodies" ne sont pas spécialisés en dispositifs
médicaux. Cela dit, le MDA veille à la
compétence des organismes qu'il accrédite. Les atouts
initiaux des deux systèmes sont donc la très bonne
connaissance du milieu pour le G-Med et la qualité des
auditeurs pour les "Notified Bodies". La seconde différence
est primordiale : Alors que le G-Med est accrédité pour
effectuer toutes les procédures de conformité, les
compétences de la plupart des "Notified Bodies" sont
limitées à un certain nombre d'annexes et de
dispositifs médicaux. Ces limitations peuvent expliquer la
politique du MDA.
Conclusion :
Sachant qu'un fabricant est libre de s'adresser à n'importe
quel organisme notifié d'un pays de l'EEE, la pluralité
des organismes notifiés permet d'accroître la
concurrence. Chaque organisme notifié doit démontrer
ses compétences et sans cesse améliorer ses
contrôles. Le problème du niveau des contrôles ne
doit pas se poser. Le débat consistant à remettre en
cause la qualité du marquage CE selon l'organisme
notifié ne sert pas l'intérêt de l'Europe. les
Autorités Compétentes sont là pour veiller au
sérieux des organismes qu'elles désignent. Les
organismes ou les pays qui "braderaient" le marquage CE seraient les
premières victimes de leur laxisme et remettraient en cause la
crédibilité du marquage CE en Europe bien sûr,
mais aussi hors d' Europe. Or, l'un des autres objectifs du marquage
CE est de valoriser les produits européens au-delà de
leurs frontières. Chaque pays doit en être
conscient.
(Retour sommaire)
|
6. Le comportement des
fabricants
|
Les Directives permettent au fabricant de choisir un organisme
notifié de n'importe quel pays de l'EEE. Face à ce
choix, on est en droit de se demander comment les fabricants
choisissent un organisme notifié :
- Un fabricant français s'adressera-t-il au G-Med
sans s'intéresser aux organismes notifiés
étrangers ?
- Selon quels critères font-ils leur choix ?
- Un fabricant anglais fait-il marcher la concurrence entre les
Notified Bodies ?
- Quel est le comportement d'un fabricant américain ou
asiatique ?
Pour répondre à ces questions qui ont certainement
des réponses diverses et variées, nous avons
établi un questionnaire
destiné à des entreprises implantées en France
et au Royaume-Uni. Un exemplaire de celui-ci est placé
à la fin du rapport.
Nous avons regroupé dans ce tableau les réponses
obtenues via les 8 questionnaires retournés (4 pour la France,
4 pour le Royaume-Uni) :
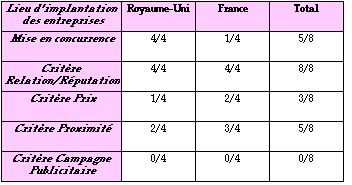
Les entreprises françaises ou anglaises qui avaient des
relations avec un organisme avant l'établissement des
directives ont garder leur partenariat. Ce partenariat était
né soit pour les questions d'homologation en France, soit pour
les questions d'assurance qualité au Royaume-Uni.
En effet, un fabricant britannique qui s'est fait certifié ISO
9001 par BSI s'est adressé à celui-ci pour obtenir le
marquage CE sachant que l'annexe II (reprise par la norme
européenne harmonisée EN 46001) se rapproche de l'ISO
9001.
La mise en concurrence :
Le comportement des fabricants français et anglais est
différent à ce sujet. Fort logiquement, les fabricants
anglais étudient plus facilement les offres des
différents Notified Bodies tandis que les fabricants
français ont plutôt tendance à choisir le G-Med
sans s'intéresser aux organismes étrangers.
De plus, il faut noter que la moitié des entreprises
britanniques sont prêtes à changer d'organismes
notifiés à tout moment si le besoin s'en faisait
sentir.
Pour ce qui est du "monopole" du G-Med, les fabricants
français n'expriment pas véritablement l'envie de voir
plusieurs organismes notifiés en France.
Les critères de choix :
Il ressort du questionnaire que les principaux critères de
choix sont la réputation et la proximité
d'un organisme notifié .
Le critère de proximité indique que le fabricant ne
s'intéressera pas aux organismes notifiés
étrangers. Pour un fabricant, la proximité est aussi
synonyme de rapidité , critère à ne pas
sous-estimé. Enfin, un autre critère, non abordé
par le questionnaire mais à prendre en considération,
reste le problème de la langue . Il est essentiel
qu'auditeurs et audités se comprennent...surtout quand on
aborde les questions techniques. Ce dernier critère peut donc
freiner certains fabricants.
Les critères de choix des fabricants non-européens
:
Les fabricants américains ou asiatiques, des
multinationales pour la plupart, qui doivent choisir un organisme
notifié européen s'adressent généralement
à leurs succursales européennes pour faire leur choix.
Cela dit, si leur lieu de production est hors d'Europe, ils
s'adressent plus facilement à des organismes notifiés
ayant des antennes dans leur pays (Tüv de Munich à San
Diego) afin de réduire les déplacements ce qui rend les
contrôles moins chers et plus rapides.
Conclusion :
Pour les entreprises implantées dans un pays
européens, le choix d'un organisme notifié
étranger ne semble pas envisagé. Les entreprises
implantées sur le sol britannique font d'abord jouer la
concurrence sur leurs terres alors que les fabricants français
se satisfont du G-Med. Les organismes notifiés s'efforcent
donc davantage "d'aller chercher" des fabricants américains ou
asiatiques. Chaque organisme notifié doit donc avoir une
dynamique commercial pour "se vendre" auprès de ces
fabricants. Rappelons que le G-Med réalise 40% de ses
contrôles hors d'Europe. Le fait d'installer des antennes dans
ces pays est un atout considérable, toujours est-il qu'il faut
en avoir les moyens...Les "petits" organismes notifiés ont-ils
un avenir ?
(Retour
sommaire)
Ce projet a proposé de faire le point sur le recours
à une tierce partie indépendante pour
l'évaluation de la conformité des dispositifs
médicaux imposée par les Directives pour
l'apposition du marquage CE. Le projet a donc confronter les
systèmes français et anglais qui diffèrent par
le nombre d'organismes habilités à réaliser ces
évaluations.
De plus, le projet s'est intéressé au comportement des
fabricants face à cette différence et plus
généralement face au choix d'un organisme
notifié en Europe. Alors que le G-Med a le monopole en
France, le MDA
fait l'unité des "Notified Bodies" au Royaume-Uni. En
instaurant une concurrence dans son propre pays, le MDA
prépare les "Notified Bodies" à la concurrence
européenne et mondiale.
Notre questionnaire rend compte de l'importance de la
proximité pour un fabricant qui recherche un organisme
notifié. Il s'adresse donc en priorité à un
organisme de son pays. La concurrence européenne apparait pour
des fabricants non européens qui veulent commercialiser leurs
dispositifs en Europe.
Au-delà de leur comparaison, ces deux choix politiques
différents traduisent parfaitement l'image du marquage
CE : Une marque communautaire qui laisse un degré de
liberté à chaque pays pour décider de la
marche à suivre pour y parvenir.
Cette liberté, on la retrouve dans les choix du fabricant
:
- Le choix de l'organisme notifié.
- Le choix de la procédure de conformité selon le
dispositif.
Ces libertés riment avec responsabilité . En
effet, la Directive spécifie la responsabilité de
chaque acteur. Cette responsabilité est essentielle quand on
arrive au niveau du patient. Etre responsable du marquage CE que l'on
appose sur un dispositif, c'est être responsable de la
sécurité du dispositif en question et donc des
patients qui seront en contact avec le-dit dispositif.
De plus le marquage CE, c'est aussi rendre les utilisateurs
responsables. En effet, le marquage CE implique des instructions
claires et précises d'utilisation .. Chaque manipulateur
(personnel médical ou techniciens) devra être
formé à l'utilisation des dispositifs par le fabricant
afin de minimiser les risques.
C'est ainsi que se décline l'attention qui doit être
apportée au patient. Penser au patient avant de penser
à soi-même...le marquage CE nous le rappelle!
(Retour
sommaire)
Accessoire : Tout article qui est destiné
principalement par son fabricant à être utilisé
avec un dispositif médical afin de permettre l'utilisation de
ce dispositif, conformément aux intentions de son
fabricant.
Autorité Compétente : C'est le ministère
de la Santé de chaque pays de l'EEE (Espace Economique
Européen). Son rôle est de mettre en application les
directives dans chaque pays. L'Autorité Compétente doit
donc :
- désigner les organismes notifiés
- assurer la mise en place du système de
matério-vigilance
- intervenir en cas de litige entre un fabricant et un organisme
notifié
Dispositif médical : Tout instrument, appareil,
équipement, matière ou autre article, utilisé
seul ou en association, y compris le logiciel nécessaire pour
le bon fonctionnement de celui-ci, destiné par le fabricant
à être utilisé chez l'homme à des fins
:
- de diagnostic , de prévention, de
contrôle, de traîtement ou d'atténuation d'une
maladie.
- de diagnostic, de contrôle, de traitement,
d'atténuation ou de compensation d'une blessure ou d'un
handicap.
- d'étude ou de remplacement ou modification de l'anatomie
ou d'un processus physiologique.
- de maîtrise de conception.
et dont l'action principale voulue dans ou sur le corps humain
n'est pas obtenue par des moyens pharmacologiques ou immunologiques
ni par métabolisme, mais dont la fonction peut être
assistée par de tels moyens.
(Retour sommaire)
Dispositifs médicaux implantables actifs : Tout
dispositif médical dépendant pour son fonctionnement
d'une source d'énergie électrique ou de toute autre
source d'énergie que celle générée
directement par le corps humain ou la pesanteur, qui est conçu
pour être implanté en totalité ou en partie, par
une intervention chirurgicale ou médicale, dans le corps
humain ou, par une intervention médicale, dans un orifice
naturel et qui est destiné à rester après
l'intervention.
Exigences Essentielles : Les dispositifs doivent satisfaire
aux Exigences Essentielles pour obtenir le marquage CE (article 3 de
la directive 93/42/CEE). Ces exigences portent sur :
- les conditions d'utilisation clinique et de sécurité
pour le patient et les utilisateurs.
- l'évaluation et l'acceptibilité des risques.
- la fonctionnalité de dispositif.
- la pérennité du dispositif.
- les conditions de protection durant le stockage et/ou le
transport.
Fabricant : C'est la personne physique ou morale responsable
de la conception, de la fabrication, du conditionnement et de
l'étiquetage d'un dispositif médical en vue de sa mise
sur le marché en son propre nom.
Organisme Notifié : Désigné par
l'Autorité Compétente de son pays, il est
accrédité pour effectuer une ou plusieurs
procédures de conformité aux exigences essentielles
décrites dans les annexes des directives. Il doit
répondre aux normes des procédures qu'il
préconise (série EN45000) . Il doit :
- être indépendant
- être impartial
- être responsable
- être techniquement compétent
- avoir les équipements adéquates
- faire voeux de confidentialité
Enfin, chaque organisme notifié possède un
numéro d'identification de trois chiffres que l'on retrouvera
sur les dispositifs qu'il certifient. Le marquage CE consiste donc en
deux lettres CE + trois chiffres (ex: CE 459 pour le G-Med).
Normes européennes harmonisées : Ces normes ont
été écrites parallèlement aux directives
dans le but de traduire les Exigences Essentielles en
spécifications techniques. A ce jour, la plupart des
procédures de conformité aux exigences essentielles
possède une norme qui la reprend (ex: l'annexe II pour le
système complet d'assurance qualité est traduite par la
Norme NF EN 46001).
(Retour sommaire)
Directive 90/385/CEE relative aux dispositifs médicaux
implantables actifs (DMIA), du 20 Juin 1990 parue au JOCE L.189 du 20
Juillet 1990.
Directive 93/42/CEE relative aux dispositifs médicaux du 14
Juin 1993 parue au JOCE L.183 du 12 Juillet 1993.
(Retour
sommaire)
MDA (Medical
Devices Agency)
"European & Regulatory Affairs" Tél : 00 44
171 972 8300
Hannibal House Fax : 00 44 171 972 8112
Elephant & Castle
London SE1 6TQ - GB
Email : mda_mail@mda.win-uk.net
Internet : http://www.open.gov.uk/mda/mdahome.htm
9 UK Notified Bodies
Amtac Certification Services Ltd
Norman Road Contact : Mr Alan Kirwilliam
Broadheath
Altrincham Tél : 00 44 161 928 8924
Cheshire Fax : 00 44 161 927 7359
WA14 4EP
BSI Product Certification
PO Box 375 Contact : Mr Doug Poole
Milton Keynes
MK14 6LL Tél : 00 44 190 831 2636
Fax : 00 44 190 869 5157
Bureau Veritas Quality International Ltd
14 Challenge House Contact : Mr Alan Butvenick
Sherwood Drive
Bletchey Tél : 00 44 190 836 6724
Milton Keynes Fax : 00 44 190 836 6725
MK3 6 DP
Inchape testing Services Ltd
Manfield Park Contact : Mr Donald Sherratt
Cranleigh
Surrey Tél : 00 44 148 326 8800
GU6 8PY Fax : 00 44 148 326 7579
Lloyd's Register Quality Assurance Ltd
Carolyn House Contact : Ms Pauline Hylton
Dingwall Road
Croydon Tél : 00 44 181 688 6883
CR0 9 XF Fax : 00 44 181 681 8146
Medicert Ltd
3 Beech Avenue Contact : Mr Alan MacGregor
Sherwood Rise
Nottingham Tél : 00 44 115 960 9857
NG7 7LJ Fax : 00 44 115 969 1788
National Quality Assurance Ltd
Gainsborough House Contact : Mr Steve Clark
Houghton Hall Park
Houghton Regis Tél : 00 44 158 286 6766
Dunstable Fax : 00 44 158 286 6700
LU5 5ZX
SGS Yarsley International Certification Services Ltd
Formal House Contact : Mr Chris Jepson
Oldmixon Crescent
Weston-Super-Mare Tél : 00 44 193 464 1608
Somerset Fax : 00 44 193 464 1609
BS24 9AL
Sira Certification Services
South Hill Contact : Mr Eric Waters
Chislehurst
Kent Tél : 00 44 181 467 2636
BR7 5EH Fax : 00 44 181 295 3005
Ministère des Affaires sociales, de la Santé et
de la Ville
Direction des hôpitaux
Bureau des dispositifs médicaux (EM1)
8, avenue de Ségur
75350 Paris 07 SP
G-Med
33, avenue du Général Leclerc
92260 Fontenay aux roses
Tél : 01 40 95 63 54
Fax : 01 40 95 62 43
(Retour sommaire)
1 - What Medical Devices of your firm are certified ?
2 - What is (are) your Notified Body(es) ?
3 - Criteria of selection :
* Reputation
: YES
NO
* Price :
YES
NO
* Proximity :
YES
NO
* Rapidity :
YES
NO
* Advertising campaign : YES NO
4 - Did you compare the different Notified Bodies ? Did you have a
difficult choice to do ? What is the main reason of your choice ?
5 - Is your choice definitive ? Why ?
6 - Are you well informed about the CE Marking ? How do you get
information ?
7 - Has the CE Marking changed your policy of production ? Why ?
8 - Other information
(Retour sommaire)