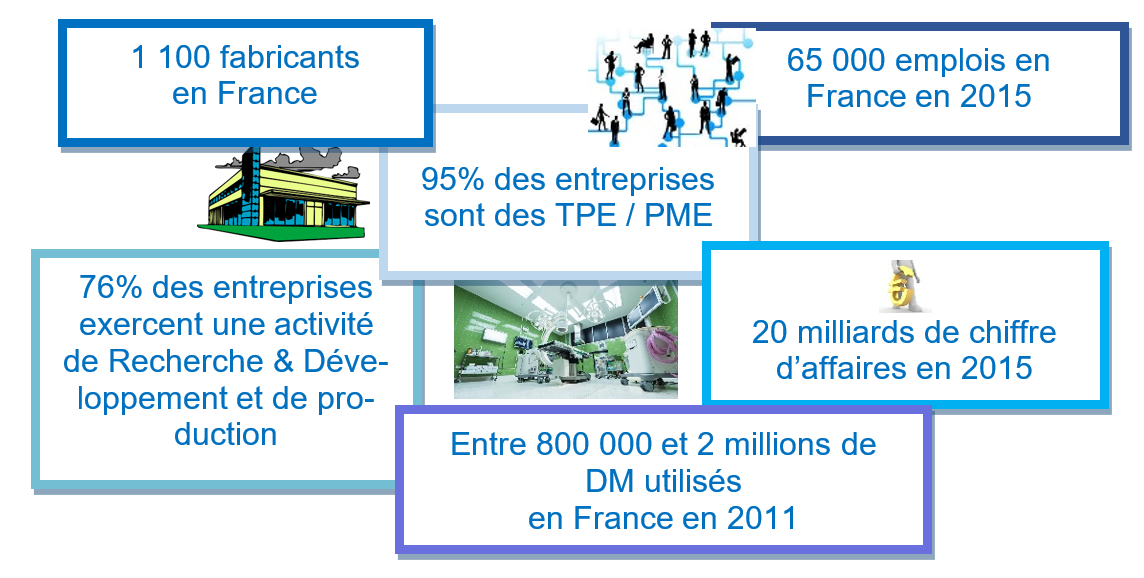
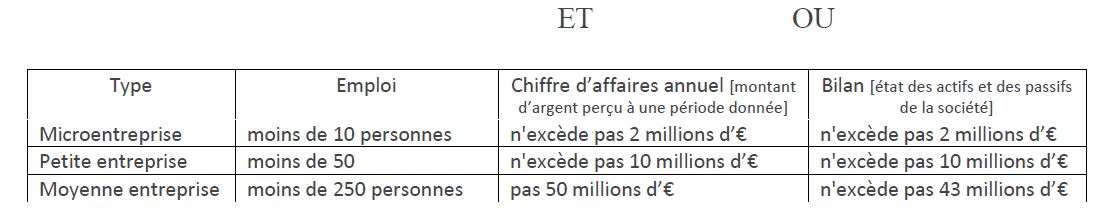
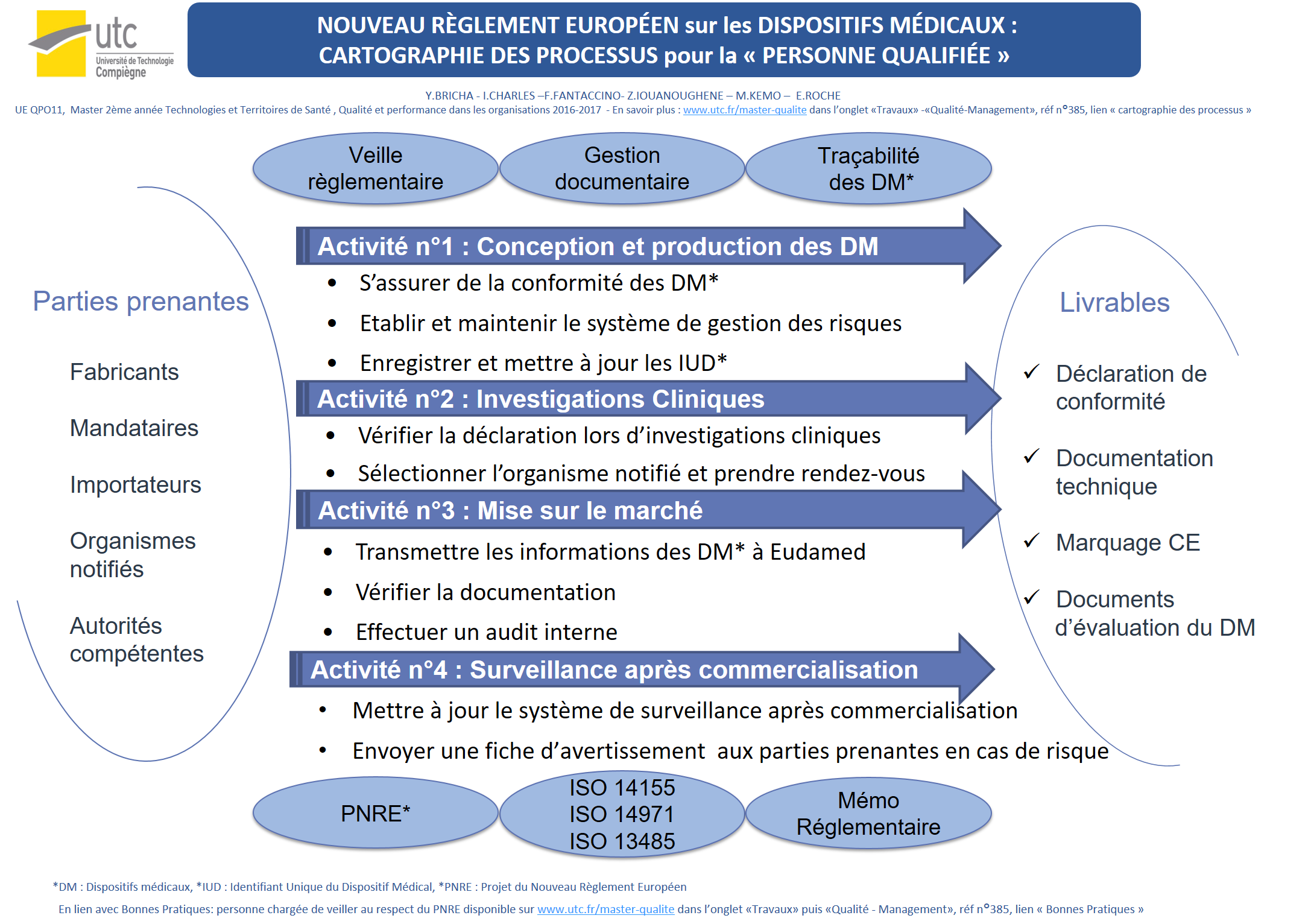
Domaine
|
Directive
90/385 =
DMIA
|
Directive
93/42 =
DM
|
Futur
Règlement
|
Champs d’application
|
DMIA
Article
1§1.
|
DM et accessoires
Article
1§1. |
Mise sur le marché
Mise à disposition sur le marché
Mise en service des DM et de leurs
accessoires
Investigations cliniques sur les DM
Article 1§1. |
Définitions
|
DM
DM actif
DMIA
Dispositif sur mesure
Dispositif à investigations cliniques
Destination
Mise en service
Article
1§2.
|
DM
Accessoire
DMDIV
Dispositif sur mesure
Dispositif à investigations cliniques
Fabricant
Destination
Mise sur le marché
Mise en service
Article 1§2.
|
Reprend ce qu’il y a précédemment avec en
plus les logiciels, les phases du cycle de vie du
dispositif, les opérateurs économiques.
Article
2§1.
|
Exclusions
|
Pas de paragraphe sur les exclusions
|
DMDIV
DMIA
Médicament
Produits cosmétiques
Sang et produits sanguins, plasma, cellules
sanguines, origine humaine
Organes, tissus et cellules d’origine
humaine
Organes, tissus et cellules d’origine
animale sauf si rendu non viable
Equipements de protection individuelle
régis par la directive 89/686.
Article
1§5et6.
|
DMDIV
Médicaments selon directives 2001/83
Médicaments de thérapie innovante selon
règlement 1394/2007
Produits cosmétiques
Sang et produits sanguins, plasma, cellules
sanguines, d'origine humaine
Organes, tissus et cellules d’origine
humaine
Organes, tissus et cellules d’origine
animale sauf si rendu non viable
Produits contenants des substances
biologiques viables, micro-organismes vivants,
bactéries, champ, virus
Denrées alimentaires.
Article
1§2.
|
Mise sur le marché
|
Prendre ses dispositions pour la mise
sur le marché et la mise en service des DMIA et
dispositifs sur mesure sans compromettre la sécurité et
la santé des patients et des tiers.
Article
2.
|
Prendre ses dispositions pour la mise sur
le marché et la mise en service des DM sans compromettre
la sécurité et la santé des patients et des tiers.
Article
2.
|
Pas de mise sur le marché ou en service si
les dispositifs ne sont pas conforme à ce texte.
Article
4§1.
|
Conformité
|
Le DM doit être conforme aux exigences
nationales adoptées selon les normes harmonisées.
Article
5§1.
|
Le DM doit être conforme aux exigences
nationales adoptées selon les normes harmonisées.
Article
5§1.
|
Le DM doit être conforme aux exigences et
prescription du règlement.
Article
6§1.
|
DMIA
|
Répondre aux exigences essentielles selon
la destination du DM.
Le fabricant doit éliminer ou réduire les
risques pour la personne qui implante.
Article
3§1.
|
Répondre aux exigences essentielles selon
la destination du DM
Le fabricant doit éliminer ou réduire les
risques, prendre les mesures de protection appropriées,
informer les utilisateurs des différents risques.
Article
3§1.
|
DM doit être conforme aux prescriptions
générales en termes de sécurité et de performances. Ne
doit compromettre l’état clinique, ni la sécurité du
patient et des autres personnes.
Le fabricant doit mettre en place un
système de gestion des risques sur l’ensemble du cycle
de vie du DM. Le fabricant doit évaluer les risques en
cas d’erreur de manipulation : caractéris-tiques
ergonomiques, environnementales et les connaissances
techniques des utilisateurs.
|
Classification des DM
|
|
Définition des critères et de 18 règles de
classification des DM.
Article
9.
|
Définition des critères et de 23 règles de
classification des DM. Précisions sur les modalités
d’application de ces règles.
Article
41.
|
Gestion des incidents
|
Des mesures sont prises pour recenser et
évaluer les données liées aux incidents : en cas
d’altérations caracté-ristiques ou des performances des
DM, d’inadéquation avec la notice. Retrait du marché du
DM par le fabricant en cas d’incident pour d’ordre
technique ou médical.
Article 8§1.
|
Idem article 8 avec ajout du critère lié
aux dysfonctionnements du DM.
Article
10 §1.
|
Les notions d’incident et incident grave
sont clairement définies.
Les notifications des incidents graves et
des mesures correctives de sécurité sont réalisées par
le fabricant via le système électronique notifié à
l’article 66b.
Pour l’analyse des incidents graves et
mesures correctives de sécurité prises : une enquête est
réalisée par le fabricant : une évaluation des risques
est faite en lien avec les organismes notifiés et les
autorités compétentes au niveau national.
Articles
2§1, 61§1 et 63§1.
|
Organisation de la vigilance et
notification des incidents et mesures prises.
|
L’état informe la commission et les autres
états sur les incidents et les mesures prises ou
envisagées.
Article
8§2.
|
Des dispositions sont prises pour informer
le fabricant et le mandataire lors d’un incident.
L’état informe la commission et les autres
états sur les incidents et les mesures prises ou
envisagées
Les évaluations sont réalisées en
collaboration avec l’autorité compétente.
Article
10 § 2 et 3.
|
Des mesures appropriées pour sensibiliser
les professionnels de santé, les utilisateurs et les
patients à signaler incident sont prises. Des moyens
pour faire ces signalements sont mis en place. Des
mesures nécessaires pour informer les fabricants en cas
d’incident et pour enregistrer les notifications sont
définies. Une évaluation de l’incident est réalisée par
le fabricant en lien avec les autorités compétentes et
les organismes notifiés.
Une évaluation par l’autorité compétente
doit être réalisée sur des critères liés aux préjudices,
aux différents risques et leurs gravités.
Le fabricant doit fournir un rapport
définitif présentant ses conclusions et les mesures
correctives réalisées. Ce rapport doit être disponible
via le système électronique.
Article
61 §3,
article 63 § 1, 2 et 4.
|
Apposition du marquage CE sur DM de classe
III
|
Procédure relative de déclaration de
conformité CE ou procédure d’examen CE de type.
Article
9§1.
|
Procédure relative de déclaration de
conformité CE selon système d’assurance qualité
Ou procédure d’examen CE de type.
Article 11§1.
|
Procédure d’évaluation de la
conformité CE selon le système de gestion qualité et de
l’évaluation de la documentation technique.
Ou procédure d’évaluation de la
conformité CE de l’examen type + évaluation de la
conformité sur la base de la vérification de la
conformité du produit.
Article
42§2.
retour
sommaire
|
Apposition du marquage CE sur DM de classe
IIa
|
|
Procédure relative à la vérification CE ou
procédure de déclaration de conformité CE.
Article
11§2.
|
Procédure d’évaluation de la
conformité selon le système de gestion de la qualité et de
l’évaluation de la documentation technique pour au moins
1 dispositif représentatif
Ou procédure d’évaluation de la
conformité sur la base de la vérification de la
conformité du produit + établir la documentation technique.
Article 42§4.
|
Apposition du marquage CE sur DM de classe
IIb
|
|
Procédure relative à la déclaration de
conformité CE selon le système de gestion de la qualité.
Ou procédure d’examen type de CE.
Ou procédure de déclaration de conformité
CE selon système d’assurance qualité de la production.
Ou procédure de déclaration de conformité
CE selon le système d’assurance qualité de produit.
Article
11§3.
|
Procédure d’évaluation de la
conformité selon le
système de gestion
qualité et de l’évaluation
de la documentation technique pour au moins 1
dispositif représentatif
Ou procédure d’évaluation de la
conformité sur la base de l’examen de type + évaluation
de la conformité
sur base de la vérification
de conformité produit.
Article
42§3.
|
Apposition du marquage CE sur DM de classe
I
|
|
Procédure de déclaration de conformité CE avant la mise
sur marché.
Article
11§5.
|
Procédure de déclaration de conformité
Union Européenne après élaboration de la
documentation technique.
Procédure particulière nécessitant
l’intervention d’un organisme notifié si DM chirurgical
stérile réutilisable ou avec des fonctions de mesurage.
Article 42§5. |
Apposition du marquage CE sur DM sur
mesure.
|
Procédure de déclaration relative aux DM à
destination particulière avec définition des critères à
respecter. Le fabricant peut charger le mandataire pour
la réalisation de cette procédure.
Article 9§2. |
Procédure de déclaration relative aux DM à
destination particulière avec définition des critères à
respecter. Le fabricant peut charger le mandataire pour
la réalisation de cette procédure.
Article 11§6. |
Procédure de déclaration relative aux DM à
destination particulière avec définition et renforcement
des critères à respecter, dont
- Nom et
adresse du fabricant
- Nom et
adresse du mandataire
- Déclaration
conformité aux prescriptions générales de sécurité
et de performance.
Article
42§7.
retour
sommaire |
Procédures réalisées par mandataire.
|
Définition des procédures pouvant être
réalisées par la mandataire sur demande du fabricant.
Article
9§3.
|
Définition des procédures pouvant être
réalisées par la mandataire sur demande du fabricant.
Article
11§8.
|
Contractualisation avec le fabricant des
tâches à réaliser par le mandataire. Le mandataire doit
s’acquitter au minimum de 8 actions liées à :
- la
vérification de la fabrication, de la conformité des
enregistrements, de la documentation technique et du
certificat CE
- la
mise à disposition et à la transmission
d’informations et de documentations
- l’information
du fabricant en cas de plainte et signalement des
professionnels, de patient ou utilisateurs relatif
aux incidents
Le mandataire peut mettre fin au mandat si
le fabricant ne respecte pas ses obligations.
Article
9§3.
|
Dispositif à Usage Unique
|
|
|
Concernant les dispositifs UU et leur
retraitement : définition des procédures d’autorisation,
des critères à respecter et des différentes obligations
qui apparaissent dans des domaines divers définis dans
cet article. Notamment, sur la traçabilité et
l’identification, la gestion des risques et la
validation des procédures inhérentes à tous les
processus y compris celui de nettoyage.
Article
15.
retour
sommaire
|
Dérogation de classification
|
|
Si un
Etat considère que la conformité du DM doit être
établie, une dérogation peut être faite pour rendre une
décision sur la catégorie du Dm ou sa classification.
Cette demande est justifiée auprès Commission
Européenne.
Article
13.
|
Sur demande justifiée d’un Etat, la
Commission, après consultation du GCDM,
détermine si produit répond ou non aux définitions de DM
ou d’accessoire de Dm et peut statuer sur l’application
des critères de classification ou sur la
reclassification d’un DM. Elle publie des actes
d’exécution pour statuer.
Articles
3§1 et 41§3.
|
Investigations cliniques
|
Notification d’une investigation clinique
aux autorités compétentes au minimum 60 jours avant le
début. Si pas de refus dans ce délai, le fabricant peut
débuter son investigation.
Article
10. |
Pour DM de classe III, un DM implantable
et/ou invasif de classe IIA et IIB : notification
d’une investigation clinique aux autorités compétentes
au minimum 60 jours avant le début. Si pas de refus dans
ce délai, le fabricant peut débuter son investigation.
Pour les autres DM, l’autorité compétente
peut donner son autorisation dès qu’il a reçu la
notification.
Dans ces deux cas, l’accord du comité
d’éthique doit être obtenu. Des procédures encadrant ces
investigations cliniques doivent être mises en place. Un
rapport sur ces investigations doit être réalisé et tenu
à disposition des autorités compétentes.
Article
15. |
Les modalités de réalisation des
investigations cliniques sont clairement définies. Elles
doivent garantir la protection des droits, la sécurité et la dignité
des personnes qui participent. Différentes conditions
doivent être remplies et répondre à des exigences des
autorités compétentes et du comité d’éthique comme avoir
le consentement éclairé, écrit, daté et signé, par la
personne qui participe ou de son représentant légal doit
être recueilli. Une organisation doit être mise en place
afin d’apporter une indemnisation en cas de dommage.
La demande d’investigation doit être
enregistrée dans le système électronique. Les évènements
indésirables occasionnés par ces investigations doivent
être enregistrés et notifiés. Des dispositions de
vigilance doivent être prises.
Articles
50 à 60.
|
Enregistrement des responsabilités de mise
sur marché
|
|
Désignation et enregistrement des personnes
responsables de la mise sur le marché d’un DM :
notification aux autorités compétentes.
Article
14.
|
Mise en
place d’un système électronique permettant la
création et l’enregistrement des coordonnées des
opérateurs économiques et la création d’un N° unique
d’enregistrement pour le fabricant.
Les coordonnées de la personne chargée de
veiller au respect de la réglementation sont
enregistrées.
Les rôles des opérateurs économiques sont
clairement définis.
Article
25.
|
Base de données Eudamed
|
|
|
Création d’une banque de données européenne
sur les DM : Eudamed.
Article
27.
retour sommaire
|
Mesures
préventives provenant d’un Etat ou la Commission
|
|
|
Si nécessaire, un Etat ou la Commission
peuvent mettre en place toutes les mesures préventives
nécessaires et justifiées pour protéger la santé et la
sécurité du patient. Notification immédiate des mesures
prises, en motivant leur décision.
Article
74.
|
Désignation des organismes notifiés.
|
Chaque Etat membre notifie aux autres Etats
ainsi qu’à la Commission les organismes désignés pour
effectuer les tâches de certification.
Article
11§1.
|
Chaque Etat membre notifie aux autres Etats
ainsi qu’à la Commission les organismes désignés pour
effectuer les tâches de certification.
- Attribution
d’un N° identification.
- Création
d’une liste des organismes notifiés avec leur N°
d’identification et les tâches pour lesquelles ils
sont notifiés
- Publication
au Journal Officiel Européen.
- Mise à
jour cette liste par la Commission.
Article
16§1. |
Une procédure de désignation et de
notification d’organismes est établie. Ceux-ci doivent
répondre à certaines prescriptions décrites dans cet
article.
- Publication
via le système électronique de la liste des
organismes avec leur N° d’identification et les
types de DM couverts.
- Mise à
jour cette liste par la Commission
- Description
des modalités de désignation et de
contrôle de cet organisme.
Articles
33 et 34.
|
Critères de désignation des organismes
notifiés.
|
Définition des critères minimaux pour
désigner un organisme notifié.
Article
11§2.
|
Définition des critères pour désigner un
organisme notifié : critères d’exclusion de certaines
professions.
Ces organismes doivent réaliser des
opérations d’évaluation et de vérification avec
intégrité professionnelle, de grandes compétences et
libres de toutes pressions et incitations pour ne pas
influencer dans son jugement.
Article
16 §2.
|
Définition des prescriptions applicables
aux organismes notifiés :
- Personnel
administratif, technique et scientifique en nombre
suffisant et avec expertise appropriée.
- Conformité
requise aux prescriptions dictées à l’annexe VI.
Article
29.
|
Dénotification d’un organisme notifié.
|
Si celui-ci ne répond pas aux critères,
retrait de sa désignation par l’Etat qui a fait la 1è
notification.
Article
11 §3.
|
Un organisme doit assurer ses tâches
notifiées par lui ou sous sa responsabilité. Il doit
disposer de personnel et posséder des moyens pour
accomplir ces tâches techniques et administratives.
Article
16 §3.
|
Si un organisme ne répond pas aux
prescriptions, ne réalise pas ses obligations ou ne met
pas en œuvre des mesures correctives nécessaires
l’autorité nationale peut suspendre, restreindre ou
retiré tout ou partie de sa désignation. Celle-ci doit
en informer immédiatement la Commission et les autres
états.
Article
36 §2.
retour
sommaire
|
Non-respect du texte règlementaire
|
|
Le fabricant doit souscrire une assurance
responsabilité civile sauf si cette responsabilité est
couverte par l’état.
Article
16§6. |
Si le fabricant ne respecte pas ce texte:
il encoure une suspension, une annulation ou une
restriction du certificat sauf s’il met des mesures
correctives en place dans un délai donné par organisme
notifié.
Article
45§3.
|
Organismes
notifiés : obligations
|
|
Personnel des organismes notifiés soumis au
secret professionnel pour tout ce qu’il apprend dans le
cadre de ses fonctions.
Article
16§7.
|
L’organisme notifié demande sa désignation
auprès de l’autorité compétente en précisant ses
activités d’évaluation et les types de DM qu’il veut
contrôler. Il doit fournir un document attestant son
respect à ce présent règlement après évaluation par un
organisme notifié d’accréditation. L’autorité nationale
contrôle les organismes présents sur son territoire.
Articles
31§2 et 35§1.
|
Non-conformité du DM
|
Si le marquage CE
est apposé à tort, l’organisme notifié prend les mesures
appropriées et informe l’autorité compétente.
Articles
13 et 14. |
Si le marquage CE est
apposé à tort : le fabricant ou le mandataire a
obligation de faire cesser l’infraction.
Si l’infraction persiste : l’Etat prend
toutes les mesures utiles pour restreindre ou interdire
la mise sur
le marché d’un DM et veiller à son retrait.
Article
18. |
Si la surveillance après commercialisation
ou la vigilance concluent à une non-conformité :
- Délai
défini pour réaliser la mise en conformité.
- Si
délai non respecté : l’Etat prend toutes les
mesures pour restreindre ou interdire la mise à
disposition du DM sur le marché.
- Notification
à la Commission et aux autres Etats via le système
électronique et sans délai.
Article
73.
|
Règles de confidentialité
|
Confidentialité des informations obtenues
dans le cadre des missions des fabricants et
mandataires.
Ne concerne pas les informations
réciproques et les diffusions de mise en garde.
Article
15.
|
Confidentialité des informations obtenues
dans le cadre des missions de toutes personnes
concernées par cette directive. .
Ne concerne pas les informations
réciproques et les diffusions de mise en garde, ni
l’obligation d’information incombant aux personnes
concernées dans le cadre du droit pénal.
Article
20.
|
- Confidentialité des informations
obtenues dans le cadre des missions de toutes les
parties concernées par l’application de ce
texte.
- Modalités d’échange
d’informations entre autorités compétentes et
Commission
- Ne concerne pas les informations
réciproques et les diffusions de mise en garde, ni
l’obligation d’information incombant aux personnes
concernées dans le cadre du droit pénal
- La commission et les états :
possibilité d’échange informations confidentielles
avec les autorités de pays tiers si des accords en
matière de confidentialité sont rédigés.
Article
84.
|
Coopération entre Etats membres.
|
|
|
Les autorités compétentes des différents
Etats coopèrent entre eux et avec la Commission.
Celle-ci veille à organiser des échanges d’information
pour une application uniforme de ce texte.
Les Etats membres participent aux mises au
point internationales et coopèrent avec les autorités
chargées de la règlementation des DM.
Article
77.
retour sommaire |