|
Avertissement
|
| Si vous arrivez directement
sur cette page, sachez que ce travail est un rapport d'étudiants
et doit être pris comme tel. Il peut donc comporter des imperfections
ou des imprécisions que le lecteur doit admettre et donc supporter.
Il a été réalisé pendant la période
de formation et constitue avant-tout un travail de compilation bibliographique,
d'initiation et d'analyse sur des thématiques associées aux
technologies biomédicales.
Nous ne faisons
aucun usage commercial et la duplication est libre. Si vous avez des raisons
de contester ce droit d'usage, merci
de nous en faire part . L'objectif de la présentation
sur le Web est de permettre l'accès à l'information et d'augmenter
ainsi les échanges professionnels. En cas d'usage du document, n'oubliez
pas de le citer comme source bibliographique. Bonne lecture... |
Référence
à rappeler :
Conséquences de l'application
de l'Arrêté du 3 Mars 2003 dans le fonctionnement d'un
service Biomédical
L. Bonifazzi, F. Ferré,
Projet DESS "TBH", UTC, 03-04
URL : https://www.utc.fr/~farges
Conséquences de
l'application de l'Arrêté du 3 mars 2003
dans le fonctionnement
d'un service biomédical
|

Luc
Bonifazzi
|

Frédéric
Ferré
|
|
RESUME
La mise en uvre par lexploitant de lobligation
de maintenance et du contrôle de qualité des dispositifs
médicaux est définie dans le décret du 5 décembre.
Il restait donc à déterminer les familles des dispositifs
médicaux concernés et à établir les modalités
de contrôle qualité de ces dispositifs.
Larrêté du 3 mars 2003 répond
pour une part à cette attente en fixant les listes des dispositifs
médicaux soumis à lobligation de maintenance et au contrôle
de qualité (interne et externe) ainsi que les dates dapplication.
A ce jour, seuls les modalités de contrôle qualité
interne et externe des mammographes analogiques ont été
publiées par lAFSSAPS. En ce qui concerne le contrôle qualité
externe, il ne peut être réalisé que par des organismes
agréés.
Une étude spécifique effectuée
auprès dune centaine dingénieurs biomédicaux met
en avant les diverses conséquences de ce texte. Les résultats
ont montré un manque de consensus sur la classe de risque des dispositifs
médicaux. Les ressources financières et humaines ne semblent
pas adaptées pour répondre aux besoins engendrés par
ce texte. Cette étude accentue également le besoin dune
politique de maintenance pour appliquer ce règlement. Larrêté
du 3 mars 2003 amène à repenser la procédure dachat
pour intégrer les coûts quil induit (maintenance, contrôle
qualité, ECME
). 80% des ingénieurs biomédicaux pensent
qu'il est nécessaire de mettre en place un programme d'assurance
qualité comprenant les acteurs médicaux et paramédicaux.
Cet arrêté cadre parfaitement
avec les exigences actuelles : la sécurité et la qualité
des soins. Il sinscrit dans la démarche qualité à
mettre en place dans les établissements de santé visant à
une évaluation permanente et une amélioration continue de
leur organisation.
Larrêté confirme également
la métamorphose du métier de technicien biomédical
et conforte lingénieur biomédical dans son rôle de
conseiller de lexploitant. Ce dernier est juridiquement responsable des
conséquences de lapplication de cette réglementation.
Mots clefs : Conséquences
- Dispositif médical - Arrêté - Décret - Maintenance
- Contrôle de qualité - Organismes agréés -
Politique de maintenance -Démarche qualité
|
ABSTRACT
Obligation to maintenance and quality control
of medical devices is defined in the decree of the 5th of December 2001.
A list of medical devices concerned by this decree needed to return. The
order of the 3rd of March 2003 published by the health ministry partially
answers this. It fixes the mandatory dates of applications and it is composed
of three lists.
The first list concerns the obligation
of maintenance, the second concerns the obligation of internal control
quality and the third the obligation of internal control quality.
Today, only the modality of control quality
concerning analogical mammographs is published. And only approved institutions
can implement them to external quality control.
A specific study among biomedical engineers
shows the various consequences of this application. The results showed
a lack of consensus on the classification of medical risk for medical devices.
Financial and human resources seem not be enough to support the requirements
of this text. This study also highlights the need of good servicing management
to comply with the regulations. In addition, the servicing could be included
in the purchase process to optimise hospital cost.
80% of bio medicals engineers think it
is necessary that a quality insurance program including medical and paramedical
actors could improve the application. The order fits perfectly with the
current requirements: security and quality of care. Process quality validates
and improves the medical organization.
The order confirms the evolution of biomedical
technician function and confirms the biomedical engineer as the directors
adviser. This latter is the only responsible person facing the legal consequences
of these regulations.
Key words : Consequences - Medical
device - Order - Decree - Maintenance - Quality control - Approved
institution - Servicing management - Quality insurance program
|
REMERCIEMENTS
Nous adressons nos plus vifs remerciements
à Monsieur Georges Chevallier, directeur du DESS "TBH", pour ses
renseignements avisés.
Nous tenons à remercier Monsieur
Gilbert Farges, enseignant chercheur à l'UTC de Compiègne,
pour les outils qualités qu'il nous a enseignés.
Nous souhaitons également remercier
pour leur contribution :
- Monsieur Julian, ingénieur
biomédical au CHU dAmiens
- Monsieur Son Luu, consultant biomédical
- Monsieur Hamon, ingénieur biomédical
au CH de Compiègne
- Monsieur Gohmari, ingénieur biomédical
au CHU de Rouen
- Monsieur Berthier, Directeur adjoint
AFSSAPS
(Direction de lévaluation
des Dispositifs Médicaux)
Enfin, nous remercions lensemble des ingénieurs
biomédicaux qui ont eu lamabilité de répondre à
notre enquête, ainsi que lensemble des acteurs qui ont participés
à cette étude.
SOMMAIRE
INTRODUCTION
PREMIERE
PARTIE : REGLEMENTATION EN VIGUEUR
I.1 RAPPEL REGLEMENTAIRE
I.2 DECRET DU 5 DECEMBRE 2001
I.3 ARRETE DU 3 MARS 2003
I.3.1 Rappel sur la classification des DM
I.3.2 Organismes Notifiés
I.4 DECISION DU 27 MARS 2003
I.4.1 Annexe de la décision du 27 mars 2003 :
I.4.2 Organismes agréés pour le contrôle qualité
externe :
I.5 NOTION DE MAINTENANCE ET DE CONTROLE DE QUALITE
I.5.1 Maintenance corrective et préventive
I.5.2 Contrôle de qualité
DEUXIEME
PARTIE : MISE EN UVRE DE LARRETE
II.1 PRINCIPAUX PROCESSUS POUR APPLIQUER LARRETE
II.2. EVALUATION DE CET ARRETE AU SEIN DES SERVICES BIOMEDICAUX
II.2.1. Elaboration de lenquête
II.2.2 Analyse et interprétation de lenquête :
II.3 BILAN
TROISIEME
PARTIE : CONSEQUENCES ET PRECONISATIONS
III.1 LISTES DES DISPOSITIFS MEDICAUX ET EVOLUTION VERS UN RSQM
III.2 POLITIQUE DE MAINTENANCE
III.3 DEMARCHE DASSURANCE QUALITE
III.3.1 Le cycle P.D.C.A
III.3.2 les référentiels
ETAPES ENVISAGEES PAR LAFSSAPS
CONCLUSION
BIBLIOGRAPHIE
ANNEXES
ANNEXE 1 : METHODOLOGIE DE LELABORATION DUNE ENQUETE :
ANNEXE 2 : ENQUETE
ANNEXE 3 : DECRET DU 5 DECEMBRE 2001
ANNEXE 4 : ARRETE DU 3 MARS 2003
Introduction
Le décret du 5 décembre 2001
et larrêté du 3 mars 2003 sont complémentaires :
Le premier impose aux exploitants de dispositifs
médicaux de mettre en uvre des maintenances et des contrôles
de qualité internes ou externes de ses dispositifs.
Le second spécifie les dispositifs
médicaux soumis à cette obligation de maintenance et de contrôle
qualité.
Qu'en est-il de leur application dans
les Centres Hospitaliers ?
Les services biomédicaux ont-ils
mis en place des organisations qui répondent aux exigences fixées
? Quelles sont leurs principales difficultés?
Lobjectif de ce dossier est donc de :
Rappeler les critères
organisationnels essentiels à mettre en place
Réaliser un
premier bilan sur lavancement des services biomédicaux
Résumer les
principales difficultés auxquelles sont confrontés les services
biomédicaux
Proposer quelques
recommandations
Première
partie : Réglementation en vigueur
I.1 Rappel réglementaire
La directive 93/42/CEE du 23 août
1993
En imposant le marquage CE, cette directive
donne le véritable départ de la notion de qualité
pour le dispositif médical. Il implique les fabricants (qui doivent
apporter la preuve de sa conformité aux Exigences Essentielles qui
repose exception faite de la Classe I, sur des certifications dOrganismes
Notifiés) mais également les exploitants qui ont une obligation
de prendre des dispositions nécessaires pour assurer en permanence
le bon fonctionnement du dispositif médical, la sécurité
des patients et des utilisateurs.
Le décret 96-32 du 15 janvier
1996
Ce décret décrit le système
de matériovigilance qui définit, notamment, la surveillance
du risque de dangerosité des dispositifs médicaux lors de
leur utilisation et l'organisation du système national de matériovigilance.
L'arrêté du 3 octobre 1995
Cet arrêté précise
les modalités dutilisation et de contrôle des matériels
et dispositifs médicaux.
Cet arrêté impose la mise
en place dans tout établissement de santé d'une organisation
spécifique pour s'assurer que tous les matériels et dispositifs
médicaux destinés à assurer l'anesthésie et
la surveillance post-interventionnelle :
Soient contrôlés
lors de leur première mise en service et lors de toute remise en
service pour s'assurer que l'installation est faite conformément
aux spécifications prévues par le participant,
Font l'objet d'un contrôle de
leur bon état et leur bon fonctionnement avant chaque utilisation
sur des patients,
Font l'objet d'une maintenance organisée,
adaptée à leurs conditions d'utilisation.
Les contrôles de mise en service, de
vérification de leur bon état et de leur bon fonctionnement,
ainsi que de maintenance doivent faire appel à des procédures
spécifiques à chaque famille de dispositifs.
Directive européenne du 30 juin
1997
La directive européenne "97/43/Euratom
" vient en complément de la directive 96/29/Euratom du 13 mai 1996
fixant les normes de base relative à la protection sanitaire de
la population et des travailleurs contre les dangers des rayonnements ionisants.
Elle comprend 16 articles construits autour
de deux principes fondamentaux de radioprotection : la Justification de
l'acte et l'Optimisation des expositions.
Le service biomédical est essentiellement
concerné par larticle 8 qui précisait déjà
les exigences suivantes :
- un inventaire à jour
des équipements radiologiques, pour chaque installation radiologique,
doit être à la disposition des autorités
compétentes.
- des programmes appropriés d'assurance
de qualité, comprenant des mesures de contrôle de qualité
et des évaluations de la dose du patient ou de l'activité
administrée, doivent être mis en uvre par l'exploitant de
l'installation radiologique et qu'un essai de réception soit effectué
avant la première mise en service des équipements à
des fins médicales et, ensuite, à ce qu'un contrôle
des performances soit réalisé régulièrement
et après chaque entretien important.
Loi du 1er juillet 1998
Cette loi N° 98-535 est relative au
renforcement de la veille sanitaire et du contrôle de la sécurité
sanitaire des produits destinés à lhomme, sur la maintenance
et le contrôle de qualité des dispositifs médicaux
Par cette loi, le Législateur transpose
en droit national les dispositions de la directive 97/43 et souhaite voir
élaborer un outil pour le maintien de la conformité des performances
du dispositif médical mis sur le marché et en service. Cette
demande se traduit dans la publication du décret du 5 décembre
2001.
I.2 Décret du 5 décembre
2001
Ce décret est relatif à l'obligation
de maintenance et au contrôle de qualité des dispositifs médicaux.
Il précise en outre la responsabilité de l'exploitant du
Dispositif Médical ainsi que son obligation de gestion et de vérification
de cette maintenance.
Définitions
Définition de lexploitant : lexploitant
dun dispositif médical est toute personne physique ou morale assurant
la responsabilité juridique de lactivité requérant
lutilisation de ce dispositif.
Définition de la maintenance : la
maintenance dun dispositif médical est lensemble des activités
destinées à maintenir ou à rétablir un dispositif
médical dans un état ou dans des conditions données
de sûreté de fonctionnement pour accomplir une fonction requise.
Les conditions de réalisation de la maintenance sont fixées
contractuellement, sil y a lieu, entre le fabricant ou le fournisseur
de tierce maintenance et lexploitant.
Définition du contrôle qualité
: le contrôle qualité dun dispositif médical est lensemble
des opérations destinées à évaluer le maintien
des performances revendiquées par le fabricant ou, le cas échéant,
fixées par le directeur général de lAgence Française
de Sécurité SAnitaire des Produits de Santé (AFSSAPS)
Le contrôle qualité est dit interne, sil est réalisé
par lexploitant ou sous sa responsabilité par un prestataire. Le
contrôle qualité est dit externe, sil est réalisé
par un organisme indépendant de lexploitant, du fabricant et de
celui qui assure la maintenance du dispositif.
Modalités de contrôle de
la qualité
Cest le directeur général
de lAFSSAPS qui définit les modalités particulières
du contrôle de chacun des dispositifs soumis au contrôle de
qualité interne ou externe. Sa décision, relative au contrôle,
est publiée au Journal Officiel de la République Française.
Concernant les dispositifs médicaux exposant les personnes à
des rayonnements ionisants, les décisions du Directeur Général
de lAFSSAPS sont prises au vu de lavis de la Direction Générale
de la Sûreté Nucléaire et de la Radioprotection (DGSNR).
Le directeur général de
lAFFSAPS fixe un référentiel de contrôle qualité
pour chaque dispositif médical comprenant :
les critères dacceptabilité
auxquels doivent répondre les performances ou les caractéristiques
des dispositifs médicaux
la nature des opérations de contrôle
à mettre en uvre pour sassurer du maintien des performances du
dispositif médical et les modalités de leur réalisation
la périodicité des contrôles
et les situations nécessitant un contrôle en dehors des contrôles
périodiques
la nature des opérations de maintenance
si le dispositif médical considéré nécessite
un nouveau contrôle en dehors des contrôles périodiques
les recommandations en matière
dutilisation et de remise en conformité « compte tenu des
dégradations ou des insuffisances de performances ou des caractéristiques
constatées »
les délais laissés à
lexploitant pour remettre en conformité le dispositif
Mise en uvre de lobligation de
maintenance et du contrôle qualité
Pour les dispositifs médicaux soumis
à lobligation de maintenance, au contrôle qualité
interne et / ou au contrôle qualité externe, lexploitant
doit :
disposer dun inventaire de
chaque dispositif quil exploite, tenu à jour, mentionnant les dénominations
communes et commerciales, le nom du fabricant et du fournisseur, le numéro
de série, sa localisation et la date de sa première mise
en service
définir et mettre en uvre une
organisation destinée à sassurer de lexécution de
la maintenance et du contrôle qualité interne ou externe des
dispositifs médicaux. Les modalités de cette organisation
sont précisées dans un document. Dans les établissements
de santé, lorganisation, adoptée après avis des instances
médicales consultatives, est portée à la connaissance
des utilisateurs. Les changements de lorganisation donnent lieu, sans
délai, à la mise à jour du document
disposer dinformations permettant dapprécier
les dispositions adoptées pour lorganisation de la maintenance
et du contrôle qualité interne ou externe, ainsi que les modalités
de leur exécution
tenir à jour un registre, pour
chaque dispositif médical, registre dans lequel sont consignées
toutes les opérations de maintenance et de contrôle qualité
interne ou externe, avec pour chacune delles lidentité de la personne
qui les a réalisées et son employeur, la date de réalisation
des opérations effectuées et la date darrêt et de
reprise dexploitation (en cas de non-conformité), la nature de
ces opérations, le niveau de performance obtenu et le résultat
concernant la conformité du dispositif médical. Ce registre
doit être conservé cinq ans après la fin dexploitation
du dispositif (sauf dispositions particulières fixées par
décision du Directeur Général de lAFSSAPS pour certaines
catégories de dispositifs)
permettre laccès aux dispositifs
médicaux à toute personne en charge de la maintenance et
du contrôle qualité
Démarche à suivre
pour le contrôles de qualité externe (CQE) :
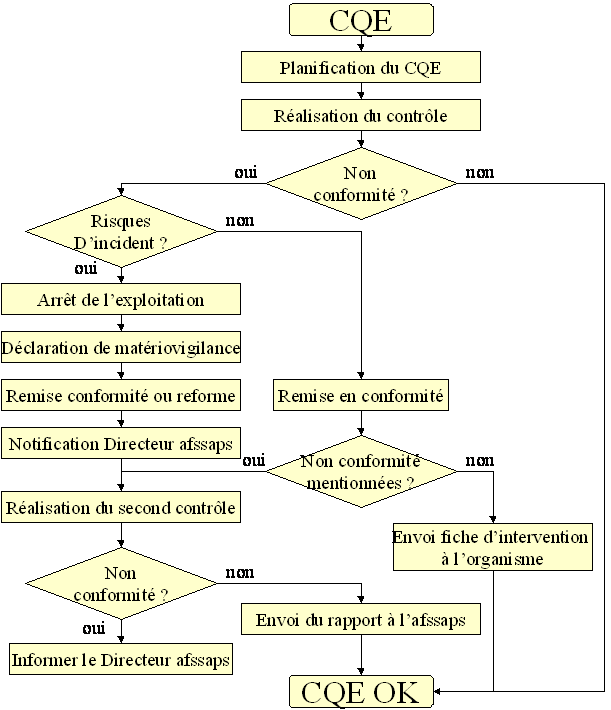
I.3 Arrêté
du 3 mars 2003
L « arrêté du 3 mars
2003 » publié au journal officiel du 19 mars 2003 fixe les
familles des dispositifs médicaux soumis à lobligation de
maintenance et de contrôle qualité ainsi que les dates dapplication.
Cette liste est résumée
dans les tableaux 1 et 2 ci-dessous :
|
Liste des Dispositifs Médicaux
soumis à lobligation de maintenance :
|
Date dapplication :
|
|
Nécessaires à la production
et à linterprétation des images radiodiagnostics
|
1 janvier 2004
|
|
Nécessaires à la définition,
planification et délivrance des traitements de radiothérapie
|
1 janvier 2004
|
|
Nécessaires à la réalisation
des actes de médecines nucléaire
|
1 janvier 2004
|
|
DM à finalité diagnostique
ou thérapeutique exposant les personnes à des RX
À lexception des dispositifs destinés
à la :
Mammographie
et ostéodensitométrie
|
1 janvier 2004
19 mars 2003
|
|
Des classes IIb et III mis en service
après le 19/03/03
Des
classes IIb et III mis en service avant le 19/03/03
|
1 janvier 2004
1 janvier 2005
|
Tableau 1 : Synthèse
de larrêté du 3 mars 2003 articles 1 2 3 et annexe I
|
Liste des Dispositifs Médicaux
soumis à lobligation
de
contrôle qualité externe et interne :
|
Date dapplication :
|
|
Nécessaires à la production
et à linterprétation des images radiodiagnostics
À lexception
des dispositifs destinés à la:
Mammographie analogique
|
Attente parution modalité de
contrôle
8 octobre 2003
|
|
Nécessaires à la définition,
planification et délivrance des traitements de radiothérapie
|
Courant 2004
(source Afssaps)
|
|
Nécessaires à la réalisation
des actes de médecines nucléaire
|
Fin 2004
(source Afssaps)
|
|
À finalité diagnostique
ou thérapeutique exposant les personnes à des RX
À lexception des dispositifs destinés
à la:
Mammographie
analogique
|
Fin 2004
(source Assaps)
8 octobre 2003
|
Tableau 2 : Synthèse
larrêté du 3 mars 2003 articles 1 2 3 et annexes
II III
I.3.1 Rappel sur la classification des
DM
On entend par Dispositif Médical
(DM) « tout instrument, appareil, équipement, matière,
produit, à lexception des produits dorigine humaine, ou autre
article seul ou en association, y compris les accessoires et logiciels
intervenant dans son fonctionnement, destiné par le fabricant à
être utilisé chez lhomme à des fins médicales
et dont laction principale voulue nest pas obtenue par des moyens pharmacologiques
ou immunologiques ni par métabolisme mais dont la fonction peut
être assistée par de tels moyens ».
Les DM sont classés selon lusage
prévu par le fabriquant ainsi que le mode daction du DM. Un appareil
peut donc avoir des classes différentes en fonction de son utilisation,
son lieu dinstallation et des compétences des utilisateurs.
Des règles de classification des
DM sont énoncées dans la Directive 93/42 (annexe IX du livre
5 bis de la Santé Publique) et sappuient sur les critères
suivants :
- le type de dispositifs invasifs
ou non-invasifs
- la dépendance dune source dénergie
- la partie du corps en contact avec le
dispositif
- la visée thérapeutique
ou diagnostique
- la durée dutilisation
- la possibilité ou non de réutilisation
En fonction des risques encourus par
les patients, les utilisateurs ou des tierces personnes, quatre classes
ont été élaborées :
- Classe I : faible degré
de risque
- Classe IIa : degré moyen de risque
- Classe IIb : potentiel élevé
de risque
- Classe III : potentiel très sérieux
de risque
Lorsque plusieurs règles sappliquent
à un dispositif médical, cest la règle correspondant
à la classe la plus contraignante qui est retenue.
Il est à noter que cest le fabriquant
lui-même qui détermine la classe de son DM. Lorsque cela est
requis, il peut faire appel à un Organisme Notifié pour confirmer
la classification de celui-ci. En cas de litige entre le fabricant et lorganisme
notifié, il incombe au Ministre chargé de la santé
de décider de la classe du Dispositif.
I.3.2 Organismes Notifiés
Les Organismes Notifiés sont des
organisations reconnues par les Etats Membres de lUnion Européenne.
Lhabilitation est réglementée.
Elle est accordée en fonction de garantie dindépendance,
de compétences, dexpériences et de moyens dans le domaine
médical.
Chaque Etat Membre désigne pour
son pays des Organismes Notifiés auxquels un numéro didentification
est attribué. Ce numéro figure généralement
au-dessous du marquage CE du Dispositif Médical.
La liste des organismes notifiés
est publiée au Journal Officiel des communautés européennes.
I.4 Décision du
27 mars 2003
Suite à l'arrêté du
3 mars 2003 fixant les listes des dispositifs médicaux soumis à
l'obligation de maintenance et au contrôle de qualité, le
directeur général de lAFSSAPS a publié le premier
référentiel de contrôle de qualité des installations
de mammographie analogique.
Les installations de mammographie analogique
sont composées de l'ensemble des dispositifs médicaux nécessaires
à la production et à l'interprétation des images de
mammographie analogique. Les contrôles sappliquent donc sur toute
la chaîne de la qualité image.
Les exploitants d'installations de mammographie
analogique doivent mettre en oeuvre cette modalité de contrôle
depuis le 8 octobre 2003. Le contrôle de qualité en mammographie
est une pratique déjà bien établie car elle bénéficiait
depuis 1998 dun référentiel. Le nouveau document, développé
par un groupe dexpert mis en place par lAFSSAPS, intègre désormais
la réglementation relative à la Matériovigilance.
I.4.1 Annexe de la décision du
27 mars 2003 :
Ce document définit les modalités
de contrôles de qualités des dispositifs médicaux nécessaires
à la production et à linterprétation des images de
mammographes analogiques conformément aux dispositions prévues
par larticle D.665-5-4 du décret du 5 décembre 2001.
Il se décompose en deux parties,
Contrôle de Qualité Externe (CQE) et Contrôle de Qualité
Interne (CQI), chacune delles précisant la périodicité
des contrôles, les opérations de contrôle à mettre
en uvre, les critères dacceptabilité des performances et
caractéristiques contrôlées et les dispositions à
prendre, le cas échéant, en cas de non conformité.
Les périodicités de ces contrôles sont présentées
dans le tableau 3.
Les CQI sont journaliers, quotidiens ou
mensuels et sont dans la majorité des cas réalisés
par les utilisateurs (manipulateurs et radiologues).
Ce référentiel, plus particulierement
les critères dacceptabilités, pourrait être revu par
lAFSSAPS car certaines installations auraient été classées
comme non-conforme avec déclaration de Matériovigilance.
|
Objet du contrôle
|
CQE
|
CQI
|
|
1- conditions de stockage des films
|
Semestrielle
|
|
|
2- chambre noire
2.1 ambiance
2.2 in actinisme
2.3 empoussièrement
|
Semestrielle
Semestrielle
Semestrielle
|
Après changement
de la lampe
|
|
3- système de développement
3.1 sensitomètre
3.2 fonctionnalité
3.3 rétention du fixateur
|
Semestrielle
SemestrielleSemestrielle
|
Quotidienne
Hebdomadaire
|
|
4- identification et état des récepteurs
4.1 identification
4.2 état et empoussièrement
4.3 étanchéité à la lumière
|
Semestrielle
Semestrielle
Semestrielle
|
En cas de changement
des cassettes
|
|
5- fonctionnalité et homogénéité des récepteurs
5.1 contact écran-film
5.2 homogénéité du parc
|
Semestrielle
Annuelle
|
|
|
6- état fonctionnel du mammographe
6.1 état général
6.2 maintien des cassettes dans potter
6.3 grille antidiffusion
6.4 système de compression
6.5 concordance des champs
6.6 alignement du faisceau avec bord du potter
|
Semestrielle
Semestrielle
Semestrielle
Annuelle
Annuelle
Semestrielle
|
Mensuelle
Mensuelle
|
|
7- exposition dans les conditions cliniques habituelles
|
Semestrielle
|
|
|
8- tension appliquée au tube
exactitude de la tension
constance de la tension
|
Semestrielle
Semestrielle
|
|
|
9- foyers
|
Annuelle
|
|
|
10- couche de demi atténuation
|
Annuelle
|
|
|
11- exposeur automatique
|
Semestrielle
|
|
|
12- qualité image
|
Semestrielle
|
Hebdomadaire
|
|
13- dose délivrée
|
Annuelle
|
|
|
14- débit du tube
|
Annuelle
|
|
|
15- cliché de référence et facteur de Bucky
|
Si changement de la
grille
|
|
|
16- salle de lecture
|
Semestrielle
|
|
|
17- négatoscopes
|
Semestrielle
|
|
Tableau 3 : Contrôle
de qualité des installations de mammographie analogique (document AFSSAPS)
I.4.2 Organismes agréés pour
le contrôle qualité externe :
En ce qui concerne le contrôle de
qualité externe, il ne peut être réalisé que
par des organismes agréés indépendants, compétents
et reconnus comme tel par les décisions de lAFSSAPS du 9 octobre
2003.
Ces organismes agréés pour
une durée de cinq ans renouvelable sont les suivantes :
AMTECH Médical
CAATS
CETE APAVE Nord-Ouest
MEDI-QUAL .
Il faut savoir que ces sociétés
sont uniquement certifiées pour la réalisation des opérations
de contrôle de qualité externe des installations de mammographie
analogique. LAFSSAPS devrait faire paraître prochainement les nouveaux
organismes agréés.
Ces sociétés ont le devoir
de remettre un rapport à lexploitant après chaque contrôle.
Ce rapport mentionne le nom de lexploitant, le dispositif contrôlé,
la nature des contrôles réalisés et les non conformités
observées .
Ces sociétés sont aussi
tenues détablir un rapport annuel dactivité (comprenant
entre autre les vérifications des dispositifs médicaux contrôlés
chez lexploitant) quils communiquent au Directeur général
de lAFSSAPS.Il est à signaler quune non conformité va de
larrêt de fonctionnement, à une déclaration de matériovigilance
et/ou à une mise en demeure de lexploitant sil na pas fait les
remises en conformité sur son dispositif.
I.5 Notion de maintenance
et de contrôle de qualité
Le décret 1154 du 5 décembre
2001 est relatif à lobligation de maintenance et de contrôle
de qualité des dispositifs médicaux. Il nous paraît
donc important dapprofondir ces deux notions.
I.5.1 Maintenance corrective et préventive
Au départ, la maintenance consistait
dans la remise en état de fonctionnement dun dispositif après
une période dite de panne. Au cours du temps, cette notion a beaucoup
évolué pour répondre aux exigences grandissantes en
termes de fiabilité et de rentabilité. Le concept de maintenance
préventive a alors été développé.
La norme AFNOR NF X 60 010 définit
la maintenance de la manière suivante : « la maintenance est
lensemble des actions permettant de maintenir ou de rétablir un
bien dans un état spécifié ou en mesure dassurer
un service déterminé ». Cette définition fait
bien la différence entre la maintenance qui a pour but de rétablir
un état, la maintenance dite corrective et la maintenance qui a
pour but de maintenir un état, la maintenance dite préventive.
Au niveau hospitalier, la maintenance
prend un sens particulier, car moyennant des compétences techniques
et organisationnelles adéquates, elle permet de prévenir
une rupture dans la continuité des soins et des dangers potentiels
pour les patients et les utilisateurs.
Nous allons maintenant détailler
les notions de maintenance corrective et de maintenance préventive.
Daprès la norme NF X 60 010,
la maintenance corrective est définie comme « lensemble des
opérations de maintenance effectuées après la défaillance
». La maintenance corrective intervient alors après la survenue
dune panne et consiste à rechercher lorigine ou cause de la défaillance
en vue de la remise en état de fonctionnement de léquipement.
Daprès la norme NF X 60 010,
la maintenance préventive est « la maintenance effectuée
selon des critères déterminés dans lintention de
réduire la probabilité de défaillance dun bien ou
la dégradation dun service rendu ». Elle a donc pour objectif
la prévention des risques de défaillances par des actions
prévues, préparées et programmées selon un
échéancier prédéterminé.
I.5.2 Contrôle de qualité
Le contrôle qualité comme
lensemble des moyens utilisés pour suivre un processus, détecter
et éliminer les anomalies (non-conformités) de celui-ci.
Faire du contrôle qualité sur un dispositif médical
consiste à évaluer les performances de celui-ci selon une
périodicité bien définie et fixée par lAFSSAPS
ou préconisée par le constructeur ou le cas échéant
prévue dans une politique dassurance qualité interne au
service.
Dans le contexte médical, la motivation
première dun contrôle qualité est de garantir la sécurité
du patient, de l'utilisateur et des tiers et de sassurer que le dispositif
considéré assure bien sa fonction pour une bonne prise en
charge du patient.
La réalisation du contrôle
qualité sinscrit parfaitement dans la réponse à ces
exigences essentielles de sécurité et de santé.
Interaction entre contrôle qualité
et maintenance préventive
Bien que la maintenance préventive
diffère du contrôle qualité, ces deux activités
n'en sont pas moins complémentaires puisque l'une peut déclencher
la réalisation de l'autre et réciproquement. Ainsi, selon
le résultat du contrôle qualité d'un dispositif médical,
le technicien peut décider de faire ou non une maintenance préventive
(dans ce cas le contrôle qualité fixe le seuil de déclenchement
dune maintenance préventive conditionnelle) ou encore, après
une maintenance préventive sur le même dispositif, le technicien
peut effectuer un contrôle qualité pour évaluer les
performances de léquipement après la maintenance et donc
la qualité de celle-ci. La maintenance et le contrôle qualité
sont 2 activités complémentaires.
Intérêts du contrôle
qualité
Dans létablissement hospitalier,
deux entités principales bénéficient directement des
résultats dun contrôle qualité pertinent :
- Le médecin garanti de
disposer dun équipement qui délivre des données correctes
(diagnostic) et/ou délivre un traitement (thérapeutique)
adéquate.
- Le service biomédical pour lequel
le contrôle qualité permet doptimiser la maintenance en permettant
de détecter une baisse de qualité (déclenchant une
maintenance), dapprécier la qualité de la maintenance corrective
réalisée aussi bien en interne par les techniciens biomédicaux
que par un prestataire externe, daméliorer les critères
de choix dun appareil (évaluation et comparaison de critères
techniques de différents dispositifs équivalents). Enfin,
l'ingénieur biomédical pourra s'appuyer sur les contrôles
effectués afin d'arrêter sa décision de réformer
ou non un appareil.
Deuxième partie : Mise
en uvre de larrêté
II.1 Principaux processus
pour appliquer larrêté
Le diagramme dIshikawa présenté
ci-dessous permet dexpliciter dans un ordre de détail croissant,
tous les moyens à mettre en uvre au niveau du service biomédical
afin de pouvoir appliquer cet arrêté. Il se décompose
en quatre 4 blocs principaux (Dispositifs, outils de maintenance et de
contrôle, management et ressource humaine) qui se subdivisent à
leur tour en plusieurs items.

II.2. Evaluation de cet arrêté
au sein des services biomédicaux
II.2.1. Elaboration de lenquête
Afin de mieux cerner et identifier les
conséquences de lapplication de cet arrêté au sein
des services biomédicaux, nous avons réalisé une enquête
auprès dune centaine dingénieurs biomédicaux à
travers toute la France (essentiellement Centre Hospitalier et CHU). Les
critères de lenquête sont décrits dans le tableau
ci-dessous.
|
Objectif de lenquête
|
Etablir un état des lieux des services
biomédicaux suite à la parution de « lArrêté
du 3 mars 2003
|
|
Echantillon
|
100 ingénieurs biomédicaux
ou responsables services biomédicaux
|
|
Type de questionnaire
|
- 1 Tableau à remplir en cochant
des cases comprenant
17
questions au choix multiple dont les 2 dernières sont des questions
ouvertes
- Fichier envoyé
par e-mail tenant sur 1 page
|
|
Déroulement de lenquête
|
- Envoi du questionnaire : 24/11/03
-
Date limite pour répondre : 15/12/03
- Relance : 05/01/03
|
Tableau 4 : récapitulatif
de létablissement de lenquête
II.2.2 Analyse et interprétation
de lenquête :
40% des services biomédicaux contactés
ont répandu à cette enquête dont 20 % de CHU et 80
% de CH. Les résultats de chaque question sont présentés
sous forme sectorielle (sauf pour les réponses aux deux dernières
questions qui sont sous forme dhistogramme) accompagnés dune analyse
plus précise.
Inventaire des DM concernés
:
Pour tenter de dimensionner limpact de cet
arrêté au sein dun service biomédical, il faut avoir
une estimation précise de la population des dispositifs médicaux
qui composent le parc déquipement. La base de départ est
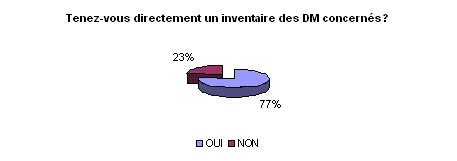
linventaire. 77% des ingénieurs biomédicaux ont un inventaire
de ces DM. Noublions pas que cest lexploitant qui est juridiquement
responsable et est tenu de disposer dun inventaire à jour des dispositifs
médicaux quil détient.
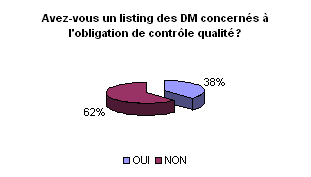
Listing des DM soumis à
lobligation de maintenance et de contrôle:
Les listes sont importantes afin de quantifier
avec précision les dispositifs médicaux affectés par
larrêté. Environ 40 % des ingénieurs ont en leur possession
des listings déquipements concernés par lobligation de
maintenance et de contrôle qualité. Dans la troisième
partie de ce rapport, nous verrons comment élaborer ces listes.

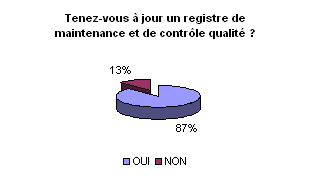
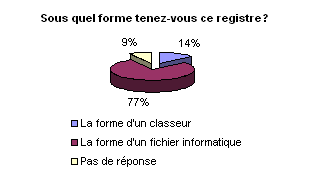
Registre de maintenance et
de contrôle qualité :
La grande majorité des ingénieurs
biomédicaux disposent dun registre pour les dispositifs médicaux
dans lequel sont consignées toutes les opérations de maintenance
et de contrôle de qualité. Ce registre peut se présenter
sous la forme dun classeur afin dêtre au plus près de léquipement
et consultable facilement par tous comme le précise le décret.
Mais en fonction du nombre de dispositifs concernés cette solution
sera limitée. Le fichier informatique, comme le confirme lenquête,
semble être la solution la plus adéquate.
Nous verrons dans la troisième
partie comment se construit un RSQM (Registre, Sécurité,
Qualité et Maintenance) selon la norme AFNOR XP S 99-171.
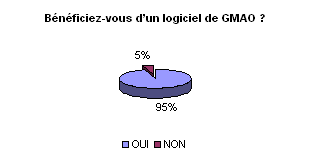
Utilisation dune GMAO :
Véritable outil pour la gestion globalisée
de la maintenance, lexploitation dune GMAO (Gestion de la Maintenance
Assistée par Ordinateur) semble incontournable, 95%des ingénieurs
en ont une. Elle permet de suivre la maintenance sous tous ses aspects
(technique, budgétaire et organisationnelle). Elle permet un gain
de temps et dobtenir une traçabilité des interventions réalisées.
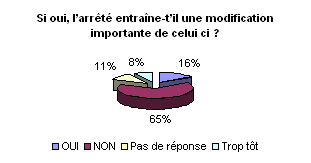
Cependant, face aux nouvelles exigences
de la réglementation, il semblerait que les GMAO ne soient pas complètement
conformes. Une évolution de celle-ci semble inéluctable mais
nengendrera pas (plus de 60% le pensent ) une modification importante
de celle-ci.
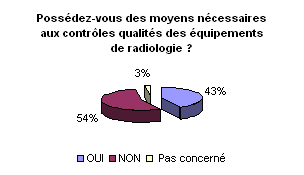
ECME (Equipements de Contrôle,
de Mesure et dEssai) :
En ce qui concerne la réalisation des
contrôles de qualité internes des équipements de radiologie,
plus de 50 % des hôpitaux possèdent des ECME, outils indispensables
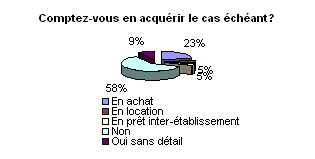
afin deffectuer les contrôles de performances. Dans les 40% à
ne pas en avoir, plus de 35% comptent en acquérir en achat, location
ou prêt inter établissement. Il faut dire que la mise en uvre
du contrôle qualité est assujettie aux publications de lAFSSAPS
fixant les modalités de contrôle. Pour linstant, il nest
apparu que les modalités de contrôle des mammographies, nous
ne connaissons pas les ECME demandés pour les autres dispositifs
médicaux dont les modalités restent à définir.
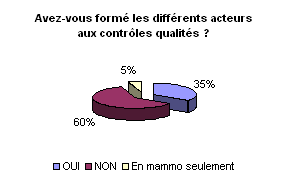
Formation des acteurs concernés
et réalisation des contrôles :
Lapplication de larrêté entraîne
une mobilisation de plus en plus importante du service biomédical
mais aussi du personnel soignant et médico-technique. Effectuer
les opérations de contrôle implique donc de disposer dun
personnel qualifié et compétent dûment formé
aux contrôles de qualité des équipements. Plus de 30%
des ingénieurs biomédicaux lont déjà fait.
La réalisation de ces contrôles nest efficace que si tous
les acteurs y adhèrent et participent activement.
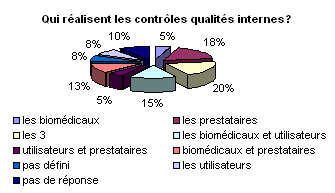
Comme en témoignent les résultats
de lenquête, plusieurs alternatives sont envisageables pour réaliser
les contrôles de qualités. Une répartition des tâches
semble indispensable entre les utilisateurs, les prestataires et les biomédicaux.
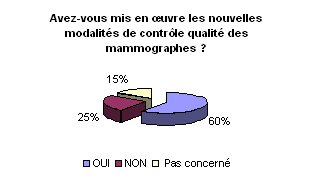
Mise en uvre des contrôles
de qualité sur les mammographes :
Le dépistage systématique
du cancer du sein en France a priorisé la parution de la décision
du 27 mars 2003 fixant les modalités du contrôle qualité
interne et externe des mammographes analogiques. Lobjectif étant
de réduire les doses au patient, obtenir une image de qualité
pour un diagnostic fiable et diminuer les rejets des films. Plus de 60%
lont bien mis en place. Rappelons que la mise en uvre de ces modalités
est rendue obligatoire depuis le 8 octobre 2003. Le contrôle qualité
sapplique sur toute la chaîne mammographique (stockage des films,
chambre noire, mammographe, cassette, développeuse, tube RX, négatoscope).
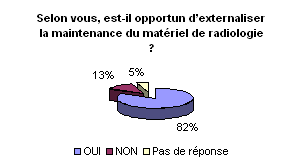
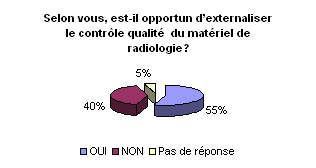
Externalisation de la maintenance
et du contrôle qualité du matériel de radiologie :
Pour garantir et optimiser sa mission en adéquation
avec les moyens et ressources dont il dispose, lingénieur biomédical
peut faire appel à lexternalisation. La tendance générale
serait comme le montrent les graphiques dexternaliser la maintenance mais
de prendre en charge le contrôle de qualité interne. Ainsi,
lingénieur biomédical peut garder la maîtrise des
performances des dispositifs médicaux et juger la qualité
de la maintenance réalisée par les sous-traitants par lanalyse
des dysfonctionnements et la traçabilité des dérives
constatées.
Bien entendu, en ce qui concerne les contrôles
de qualité externe, ils ne peuvent être faits que par des
organismes extérieurs agrées par lAFSSAPS.
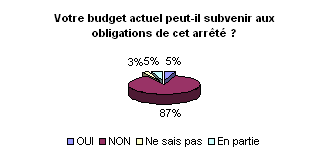
Evaluation de la maintenance
et du contrôle qualité pendant les procédures dachat
:.
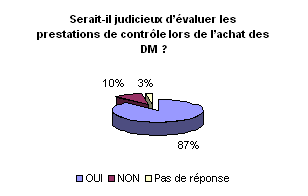
Selon 87% des ingénieurs sondés,
larrêté du 3 mars 2003 amène à repenser la
procédure dachat lors des choix dacquisition des dispositifs médicaux.
A lévidence, le coût de la maintenance et du contrôle
qualité sont des critères à prendre en compte pour
évaluer une offre dachat. Les coûts induits par larrêté
peuvent par exemple être intégrés dans des tableaux
prévisionnels dinvestissement et dans les cahiers des clauses particulières.
Lors de la révision des contrats, il sera important de dissocier
la maintenance du contrôle de qualité.
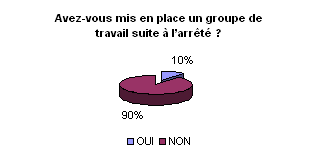
Mise en place dun groupe
de travail et présentation dune politique de maintenance :
Le service biomédical est amené
de plus en plus à travailler avec les services cliniques et médico-techniques
quant à la réalisation de certains contrôles de qualité.
Seulement 10% des ingénieurs interrogés ont déjà
mis en place un groupe de travail.
Pour déterminer les ressources
nécessaires et permettre une synergie, la mise en place dun groupe
de travail pluridisciplinaire est indispensable.
Cette approche organisationnelle passe
par une politique de maintenance, clairement définie et validée
par les instances. Lingénieur biomédical est la personne
la plus à même pour mener à bien cette organisation
et élaborer ce document (plus de 30 % lont déjà rédigé).
La politique de maintenance sera abordée
dans la troisième partie.

Programme dassurance qualité
:
La plupart des ingénieurs pensent quune
démarche dassurance qualité est nécessaire pour lapplication
satisfaisante de ce texte réglementaire. La mise en uvre de cet
arrêté amène la mise en place dun programme dassurance
qualité au sein du service biomédical, il appartient donc
à lingénieur biomédical dorganiser avec sa direction
lapplication de cette réglementation et dimpliquer tous les acteurs
concernés dans la réalisation des contrôles . Plusieurs
solutions peuvent être envisagées pour aider lingénieur
biomédical comme par exemple lutilisation du « Guide des
bonnes pratiques biomédicales ».
Une démarche dassurance qualité
peut permettre danticiper les exigences des prochaines réglementations.
Elle est également seule garante de lengagement de la direction
et de la pérennisation de lorganisation.

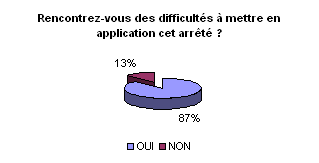
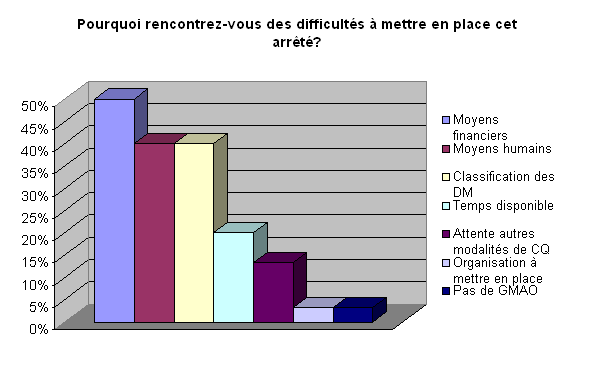
Difficultés rencontrées
quant à lapplication de cet arrêté et les raisons:
Selon la majorité des ingénieurs,
il semble difficile de mettre en uvre cet arrêté dans toute
son ampleur dun point de vue organisationnel et financier.
L approche budgétaire consisterait
à analyser les coûts directs et indirects : contrats de maintenance
des DM affectés par larrêté , coûts des ECME
(achat, calibration
), temps nécessaire à la réalisation
des contrôles, formations (techniciens et utilisateurs) et temps
dindisponibilité du matériel.
Dans lensemble, les moyens humains et
financiers semblent insuffisants. Lencadrement des crédits dexploitation
reste une constante particulièrement contraignante. De plus, les
nouvelles modalités de contrôle qualité nétant
pas encore apparues, il semble difficile destimer les coûts à
long terme.
Une autre difficulté rencontrée
selon lenquête est lidentification de la classe de risque IIb et
III du dispositif médical. Comme nous lavons vu dans le chapitre
précédent, le DM peut se classer en classe I, Classe IIa,
Classe IIb ou Classe III. Des règles de classification des DM sont
énoncées dans la Directive 93/42. Mais cette classification
nest pas aisée car elle est sujette à de nombreuses interprétations.
Beaucoup dexploitants souhaiteraient être davantage aidés
par exemple par une lettre ou circulaire. Néanmoins, il est à
noter que cest le fabriquant qui a la responsabilité de fixer la
classe de son DM et quun guide « lignes directrices » est
en cours délaboration au niveau européen ainsi quau niveau
de lAFSSAPS.
Quant à lapproche organisationnelle,
elle va permettre de définir les ressources matérielles et
personnelles. La mise en place dun tel système nest efficace que
si tous les acteurs y adhèrent et participent activement à
son fonctionnement.
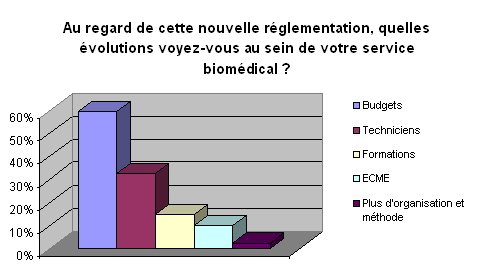
Evolution au sein des services
biomédicaux :
La majeure partie saccorde à dire
que la montée en puissance de cet arrêté par la parution
dautres modalités de contrôle qualité des dispositifs
médicaux engendrera une augmentation des ressources humaines et
financières. Le service biomédical voit en effet arriver
une augmentation de ses activités de formation de part la
sensibilisation du personnel au contrôle qualité. Les moyens
matériels devront suivre cette évolution. L application
de ce texte réglementaire tend aussi vers une nouvelle organisation
à mettre en place où lingénieur biomédical
se place en tant que chef de projet au sein dun groupe pluridisciplinaire
pour la mise en place par exemple de contrôle qualité. Il
devra pour cela disposer de tous les moyens nécessaires à
la mise en uvre de ces contrôles avec un engagement fort de la direction,
cet aspect sera développé dans la troisième partie.
II.3 Bilan
Le bilan de cette enquête est très
positif. En effet, en plus du taux de participation relativement correct,
les résultats nous ont permis de mettre en évidence certaines
missions importantes mais difficiles à maîtriser.
En outre, cette étude nous a permis
daborder toutes les conséquences de larrêté sur le
service biomédical. Lanalyse de la situation a constitué
une base solide et un apport de connaissance indispensable pour poursuivre
notre projet.
Dans la troisième partie de ce
rapport, il conviendra donc de fournir des éléments et informations
nécessaires aux services biomédicaux pour améliorer
lapplication de cet arrêté.
Troisième partie
: Conséquences et préconisations
Lenquête nous a montré que
lapplication de larrêté du 3 mars 2003 entraîne des
changements non négligeables au sein des établissements de
santé et plus particulièrement au niveau du service biomédical.
La parution dautres modalités de contrôle et de nouveaux
textes réglementaires par lAfssaps semblable à larrêté
du 3 mars 2003 vont sappliquer à dautres dispositifs médicaux
mobilisant de plus en plus le service biomédical.
Les répercussions de ces textes
réglementaires peuvent être dordre financier (coût
des contrôles obligatoires, contrats de maintenance, achat dECME,
recrutement de techniciens
) et organisationnel (temps à dégager
pour réaliser et gérer les contrôles, formations des
utilisateurs,
). Pour faire face à cette évolution, le service
biomédical doit principalement entreprendre de :
Quantifier des DM concernés
Elaborer une politique de maintenance
Mettre en oeuvre un programme dassurance
qualité
Dans cette partie, nous développerons
donc ces trois points cruciaux pour le service biomédical.
III.1 Listes des Dispositifs
Médicaux et évolution vers un RSQM
Afin de connaître limpact dans un
service biomédical, la première étape consiste à
estimer lensemble du parc des dispositifs médicaux affectés
par larrêté. Laudit initial est fondamental. Il permet de
dresser un état des lieux en établissant les listes des dispositifs
médicaux concernés et connaître alors la charge de
travail engendrée et les actions à entreprendre.
Il semble donc souhaitable délaborer
dans un premier temps les listes suivantes :
Liste des dispositifs médicaux
de classe IIb et III
Liste des dispositifs médicaux
nécessaires à la production et à linterprétation
des images de radiodiagnostic
Liste des dispositifs médicaux
de radiothérapie
Liste des dispositifs médicaux
de médecine nucléaire
Liste des dispositifs médicaux
à rayonnements ionisants
Ces listes pourront être réalisées
à laide de linventaire à jour des dispositifs médicaux
de létablissement. Par lintermédiaire du logiciel de GMAO,
on pourra procéder à lexportation du parc déquipement
au format de fichier informatique EXCEL par exemple pour obtenir un tableau
regroupant pour chaque dispositif les informations pertinentes.
Ces informations seront ensuite à
comparer avec les contrats de maintenance existants et les interventions
réalisées en interne.
Quatre points essentiels sont à
signaler :
A lheure actuelle, pour déterminer
la classe des dispositifs médicaux, on doit utiliser lannexe IX
du livre V bis du code de la santé publique, récupérer
le certificat de marquage CE ou contacter le fabriquant.
Lexploitant peut exclure des dispositifs
médicaux pour lesquels il est en mesure de justifier quune maintenance
est inutile en raison de leur conception ou de leur destination.
Les dispositifs médicaux nécessaires
à la définition, à la planification et à la
délivrance des traitements de radiothérapie sont gérés
par les radiophysiciens. Ces derniers sont responsables des doses administrées
au patient et réalisent déjà des contrôles de
qualité. Ces derniers sont également impliqués dans
les installations nécessaires à la réalisation des
actes de médecine nucléaire. Lingénieur biomédical
est donc amené à travailler en étroite collaboration
avec le radiophysicien.
Pour les installation de radiologie
conventionnelle, lingénieur biomédical et la personne compétente
en radioprotection (PCR) peuvent travailler ensemble.
GMAO et RSQM
Suite aux nouvelles exigences de ces textes
réglementaires, lévolution des logiciels de GMAO (Gestion
de la Maintenance Assistée par Ordinateur) semble inéluctable
pour quils puissent suivre les nouveaux besoins techniques et organisationnels.
Les GMAO devront prendre en compte entre autres :
Le contrôle qualité
interne (planification, enregistrement du résultat du contrôle
)
Le contrôle qualité externe
(planification, enregistrement résultat du contrôle
)
La classe de risque du dispositif médical
Le partage de linformation
Lobjectif de la norme AFNOR XP S 99-171
de décembre 2001 est de proposer la mise en uvre dun registre
pour chaque dispositif médical affecté par la réglementation
en vigueur. Ce registre pourra sintégrer dans une GMAO.
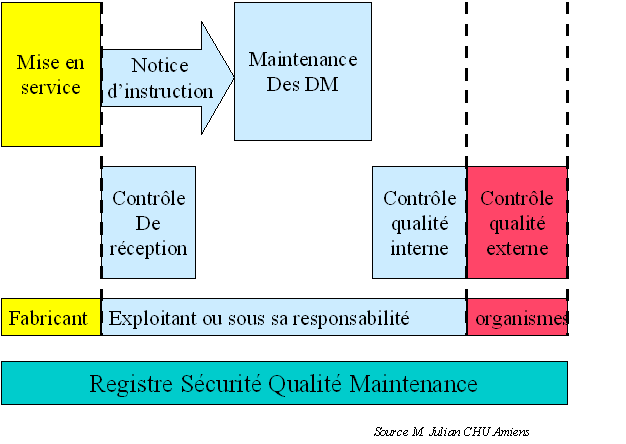
Le RSQM (Registre Sécurité
Qualité Maintenance) permet de garantir lenregistrement des données
des opérations de sécurité, de maintenance et de contrôle
qualité tout au long de la vie du dispositif. Cette traçabilité
prend en compte toutes les opérations depuis linstallation du dispositif
médical jusquà sa réforme et doit être conservé
5 ans après la fin de vie du dispositif.
Le RSQM intègre donc les informations
relatives à la réglementation et à la normalisation
en matière de maintenance pour chaque dispositif médical
et pour chaque opération prévue. Le support dinformation
peut être indifféremment réalisé par des registres
papiers à travers des classeurs ou par des enregistrements informatiques
dans un logiciel spécifique ou dans une GMAO.
Le registre doit être productible
rapidement lors de la visite des organismes de contrôle. Il doit
donc être toujours accessible ou être imprimable rapidement.
Le registre est soit lélément
unique de suivi du dispositif soit un élément dune GMAO
qui devra alors proposer déditer pour chaque dispositif affecté
un RSQM .
Le registre se compose de différentes
rubriques ou fichiers qui permettent la description du dispositif médical
et le suivi des opérations suivantes : mise en service, maintenance
préventive, maintenance corrective, contrôle qualité,
contrôle de sécurité, dans certains cas les dérogations
demploi, de réforme ou du retrait du service.
Lexploitant doit donc assurer la continuité
des informations pendant la durée de vie du dispositif. Il doit
avoir mis en uvre ou faire mettre en uvre les moyens pour garantir le
suivi la mise à jour et le transfert des données du registre
RSQM et ceci dans tous les cas possibles.
La gestion du registre RSQM doit reposer
sur une organisation, une structure définie par létablissement
et décrite dans une procédure.
III.2 Politique de maintenance
Une politique de maintenance clairement
définie répond parfaitement à lobligation mentionnée
dans le décret 1154 article D. 655-5-5 ainsi quau référentiel
GFL (Gestion des Fonctions Logistiques) du manuel daccréditation
de lANAES (Agence Nationale dAccréditation et dEvaluation en
Santé). LAfssaps devrait publier prochainement les autres modalités
de contrôle qualité, une politique de maintenance peut alors
permettre danticiper les éxigences de ces futurs référentiels.
Lingénieur biomédical est
la personne la plus à même pour élaborer la politique
de maintenance mais elle doit être validée par la direction
et les instances de létablissement ainsi quêtre présentée
aux services de soins ou médico-techniques. La politique de maintenance
doit décrire lorganisation du service biomédical : définition
des responsabilités, la qualité et la formation du personnel,
la gestion des demandes dintervention et leur enregistrement.
Suite au décret et à larrêté,
elle doit préciser comment est organisée la maintenance corrective,
la maintenance préventive mais également les contrôles
de qualité.
En effet, ces activités peuvent
être :
externalisées au constructeur
ou à une société de tierce maintenance.
réalisées en partenariat
avec les techniciens biomédicaux.
réalisées en interne par
les techniciens biomédicaux et/ou les utilisateurs.
La politique de maintenance va permettre
au service biomédical de définir son périmètre
daction. Par exemple, la maintenance des modalités dimagerie médicale
étant souvent sous traitée, les contrôles de qualité
internes peuvent être réalisés par le service biomédical
afin de juger de la qualité de la maintenance réalisée
et de mesurer les performances des équipements. Lexploitant peut
aujourdhui réaliser la maintenance et le contrôle de qualité
mais dans ce cas il est « juge et partie ».
Lorganisation mise en place sera
donc différente suivant létablissement. En fonction de la
solution qui sera retenue, les processus et les moyens à mettre
en uvre ne seront pas les mêmes.
Base de la gestion organisationnelle, la
politique de maintenance est donc primordiale pour déterminer l'organisation
et l'évolution du service biomédical. Elle doit prendre à
la fois en compte les caractéristiques de l'établissement
et y intégrer les objectifs de progrès pour le service. Ces
objectifs doivent être clairs, mesurables et définis avec
léquipe de maintenance.
Cette politique va permettre de définir
les moyens mis en uvre pour répondre aux exigences des services
de soins et médico-techniques afin d'assurer la sécurité
et la fiabilité optimale de fonctionnement des dispositifs médicaux.
La formation est donc une composante importante
de la politique qualité :
-
Formations des utilisateurs afin d'assurer
la sécurité au niveau de lutilisation des dispositifs médicaux.
Il est également souhaitable de prévoir des formations au
contrôle de qualité.
-
Formation des techniciens biomédicaux
afin d'assurer la qualité de la prestation biomédicale et
son étendue. Les formations des techniciens biomédicaux sont
très importantes afin que ces derniers puissent actualiser leurs
connaissances biomédicales et compétences techniques. Ne
pas former les techniciens biomédicaux reviendrait à limiter
leur champs daction à la gestion administrative et technique.
Pour définir la politique de
maintenance d'un Service Biomédical, il faut connaître les
moyens humains et matériels dont on dispose pour intervenir sur
les dispositifs médicaux, ainsi que les contrats de maintenance
existants. Cette situation n'est pas figée, elle évoluera
dans le temps en fonction :
Des nouvelles compétences
Des nouveaux dispositifs
Des nouveaux contrats
Des nouveaux moyens financiers, humains,
matériels
Des nouveaux objectifs
III.3 Démarche dassurance
qualité
Les services biomédicaux hospitaliers
doivent répondre à de nombreuses exigences réglementaires,
aux attentes des clients des services cliniques, médico-techniques
et administratifs. Pour cela, il leur est nécessaire de maîtriser
lensemble des processus qui contribuent à produire une prestation
de qualité reconnue par leurs clients.
La mise en uvre dune démarche
qualité au sein dun service biomédical est nécessaire,
voire indispensable aujourdhui afin de répondre à une évolution
permanente de la réglementation.
Dailleurs certains ingénieurs
biomédicaux ont déjà participé à la
mise en place dune démarche dassurance qualité au niveau
des services dimagerie médicale.
LAssurance de la qualité suivant
la norme ISO 8402 correspond à :
« L'ensemble des activités
préétablies et systématiques mises en uvre dans le
cadre du système qualité et démontrées en tant
que de besoin pour donner la confiance appropriée en ce qu'une entité
satisfera aux exigences de la qualité ».
Il faut préétablir ce que
l'on doit faire, le faire et apporter la preuve que cela a été
fait.
Nous présenterons dans les paragraphes
suivants un outil daide à la mise en place dune démarche
dassurance qualité ainsi que les principaux référentiels
afin de mener à bien les missions du service biomédical.
III.3.1 Le cycle P.D.C.A
La qualité est un processus damélioration
continue basé sur les principes de la roue de DEMING couramment
appelé Cycle P.D.C.A. (Plan : planifier, Do : faire, Check
: vérifier, Act : agir)
Ce cycle sapparente à la planification,
la mise en uvre, à la maîtrise et à lamélioration
continue des processus du système qualité.
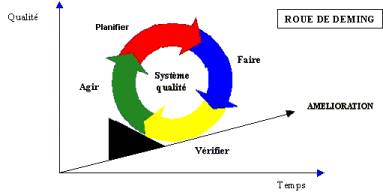
La roue de Deming se décompose
en quatre phases :
Planifier : établir les
objectifs et les processus nécessaires.
Cette phase est nécessaire pour
mettre en place une stratégie « ambitieuse et réaliste
» dans chaque processus.
Faire :réalisation des processus.
Cette phase correspond à la mise
en place des dispositions présentées dans les plans dactions.
Vérifier : mesure et évaluation
des résultats obtenus.
Cette phase correspond à la vérification,
à lévaluation des résultats et des progrès
obtenus.
Agir : actions daméliorations
envisagées
Cette phase correspond à la mise
en place dactions correctives pour améliorer les processus
III.3.2 les référentiels
Laccréditation
Laccréditation est une démarche
obligatoire pour tous les établissements de santé.
L'A.N.A.E.S. (Agence Nationale d'Accréditation
et d'Evaluation en Santé) est la structure compétente pour
gérer la procédure d'accréditation prévue par
l'ordonnance du 24 avril 1996 : « procédure d'évaluation
externe à un établissement de santé, effectuée
par des professionnels, indépendante de l'établissement et
de ses organismes de tutelle, évaluant l'ensemble de son fonctionnement
et de ses pratiques. Elle vise à assurer la sécurité
et la qualité des soins donnés au patient et à promouvoir
une politique d'amélioration continue de la qualité au sein
des établissements ».
Mais le manuel daccréditation
qui accompagne cette démarche a pour objectif lamélioration
continue de lensemble de létablissement, toutes les missions dun
service biomédical ne sont donc pas prises en considération
et évaluées.
la certification
La certification ISO 9000 (version
2000) est une démarche volontaire et elle est le fruit d'un travail
reconnu par un organisme officiel suivant un référentiel
international. C'est la satisfaction de voir déclaré atteint
un objectif fixé par une entité compétente. Mais c'est
aussi la garantie de la mise en uvre d'une organisation adaptée
qui formalise les relations internes du type "client/fournisseur".
La certification est la concrétisation
d'une démarche qui prouve la qualité d'un service.
Le seul changement significatif qu'apporte
l'édition 2000 par rapport à la version de 1994 est l'abandon
de l'approche procédurale en matière de gestion (qui définit
la manière dont vous contrôlez vos activités) au bénéfice
d'une approche fondée sur les processus (qui focalise sur ce que
vous faites).
Pour le service biomédical, la certification
peut être aussi, et surtout, un outil de management et d'amélioration
des pratiques. Mais elle réclame des efforts humains importants
et un coût financier pour sa mise en place.
le guide des bonnes pratiques
biomédicales
Le guide des bonnes pratiques biomédicales
en établissements de santé apporte les éléments
de réponse pour atteindre les objectifs et remplir les missions
dun service biomédical hospitalier. De plus, ce référentiel
prépare les services biomédicaux aux exigences dobligation
de maintenance de traçabilité et de contrôle qualité
induites par le décret du 05 décembre 2001.
La mise en uvre du Guide des bonnes pratiques
biomédicales est peut être la solution la plus adéquate
car elle repose sur des items fonctionnels et organisationnels. Ce référentiel
est le reflet de la profession biomédicale.
Etapes envisagées par
lAFSSAPS
LAFSSAPS a mis en place les étapes
suivantes pour permettre la mise en uvre progressive de larrêté
:
-
la mise en uvre dun arrêté
fixant la composition du dossier de demande dagrément des organismes
de contrôle de qualité externe qui a été publié
dans le Journal Officiel du 19 mars 2003.
-
la finalisation du référentiel
de contrôle qualité externe des installations de radiothérapie
est prévue courant 2004. En ce qui concerne le contrôle de
qualité interne, il a été soumis à la Direction
Générale de la Sûreté Nucléaire et de
la Radioprotection (DGSNR) et à lInstitut de Radioprotection et
de Sûreté Nucléaire (IRSN) en janvier 2003.
-
En ce qui concerne le contrôle
de qualité des dispositifs médicaux de diagnostic, il a été
décidé de mettre en place des groupes de travail :
-
groupe concernant les scanographes.
-
groupe concernant les dispositifs médicaux
de radiologie dentaire.
-
groupe concernant les ostéodensitomètres
(le référentiel devrait être publié assez rapidement
afin dêtre en phase avec la campagne de dépistage de lostéoporose
en préparation au ministère de la santé).
-
groupe concernent les dispositifs médicaux
de radiologie (scénario pas évident vu la diversité
des dispositifs : équipement avec ou sans scopie, appareil mobile,
table télécommandée
).
Enfin, une réflexion doit également
être menée dans les années à venir sur :
-
la publication dune circulaire dapplication
du décret du 5 décembre 2001 et de larrêté
du 3 mars 2003.
-
lélaboration dun guide de bonnes
pratiques en obligation de maintenance et de contrôle de la qualité
des dispositifs médicaux
-
lélaboration dune liste des
dispositifs médicaux non radiogènes devant faire lobjet
dun contrôle de la qualité et létablissement du calendrier
de mise en uvre.
-
la réalisation dun bilan sur
la mise en uvre de la réglementation et élargissement de
larrêté afin de créer un cadre organisationnel et
réglementaire uniforme pour lensemble des dispositifs médicaux.
Conclusion
Cette étude nous a permis de faire
ressortir les différentes répercussions au sein du service
biomédical quant à lapplication de larrêté
du 3 mars 2003.
La mise en uvre de la maintenance étant
une activité déjà établie au niveau des services
biomédicaux, larrêté nentraîne pas de grande
modification à ce niveau. Elle ne fait que confirmer la métamorphose
du métier de technicien biomédical qui glisse progressivement
vers une fonction essentiellement préventive avec une part de plus
de plus importante de gestion.
En ce qui concerne les contrôles
de qualité internes, une répartition des tâches avec
les utilisateurs semble inévitable. Dailleurs, une réflexion
pourrait être menée sur la formation de Personne Compétente
en Contrôle Qualité .
Ces textes réglementaires associés
aux parutions des modalités AFSSAPS impliquent de définir
la responsabilité, la qualité et la formation du personnel
pour la réalisation de ces contrôles. Le ministère
de la santé et lAFSSAPS à travers cette réglementation
visent implicitement la mise en uvre dune véritable politique
de maintenance clairement définie, validée par les instances
et portée à la connaissance des utilisateurs.
Interlocuteur idéal pour les échanges
et la communication entre les différents intervenants concernés
par larrêté, lingénieur biomédical avec le
soutien de la direction se place comme chef de file dun groupe de compétences
multidisciplinaires pour la mise en uvre de ces textes réglementaires.
Une démarche dassurance qualité devient alors indispensable
pour relever ce défi. En effet, lassurance qualité est seul
garant de la pérennité et de la reproductibilité du
processus mis en place.
Enfin, cet arrêté cadre parfaitement
avec les exigences actuelles : la sécurité et la qualité
des soins. Il sinscrit dans la démarche qualité à
mettre en place dans les établissements de santé visant à
une évaluation permanente et une amélioration continue de
leur organisation.
Bien que le ministère de la Santé
veuille limiter les conséquences de cet arrêté ( les
parutions actuelles ne concernent que les installations pour lesquelles
une organisation était déjà en place), dans les années
à venir, des textes réglementaires vont sappliquer à
dautres dispositifs médicaux mobilisant de plus en plus le service
biomédical mais aussi le personnel médical. Les établissements
de santé pourront-ils alors allouer les moyens humains et financiers
nécessaires à la mise en oeuvre de ces textes ?
Bibliographie
Exigences réglementaires
Arrêté du 3 mars
2003 fixant les listes des dispositifs médicaux soumis à
lobligation de maintenance et au contrôle de qualité mentionnées
aux articles L.5212-1 et D.665-5-3 du code de la santé publique,
NOR : SANP0320928A,
Décret n° 2001-1154 du 5
décembre 2001 relatif à lobligation de maintenance et au
contrôle de qualité des dispositifs médicaux prévus
à larticle L.5212-1 du code de la santé publique (3ème
partie : Décrets), JORF n° 284 du 7 décembre 2001, NOR
: MESP0123968D
Loi n°98-535 du 1er juillet 98 J.O.
du 2 juillet 1998 relatif au renforcement de la veille sanitaire et contrôle
sécurité sanitaire des produits destinés à
lhomme.
Directive 97/43/EURATOM du Conseil du
30 juin 1997 relative à la protection sanitaire des personnes contre
les dangers des rayonnements ionisants lors dexpositions à des
fins médicales.
Décisions du 27 mars 2003 de
lAFSSAPS fixant les modalités du contrôle de qualité
des installations de mammographie analogique, NOR SANM0321133S.
Directive 93/42/CEE du Conseil du 14
juin 1993 relative aux dispositifs médicaux (JO CE L-169 du 12 juillet
1993)
Ouvrages
«50 questions sur la matériovigilance»
I. LUCAS-BALOUP édition SCROF sept.96
«Guide des Bonnes Pratiques Biomédicales
en Etablissement de Santé» G. FARGES, G. WAHART, J. DENAX,
H. METAYER et 45 co-auteurs,
«Maintenance et contrôle
qualité : comment anticiper lapplication du décret n°2001-1154»
article revue RBM news 2002 n°23 de E. PELTIER-F. PRODHOMME-G.FARGES-JP
CALISTE
Norme AFNOR NF XPS 99-171 « Modèle
et définition pour létablissement et la gestion du Registre
Sécurité, Qualité et Maintenance dun D.M. »
édition AFNOR déc.2001
« Mise en situation au métier
dIngénieur Biomédical et contrôle qualité en
radiologie » rapport de stage E. BERENGER session DESS TBH 2002-2003
« Elaboration dun outil dauto
diagnostic du service biomédical afin dévaluer ses prestations
face à ses obligations » rapport de stage R. GIGLEUX
session DESS TBH 2001-2002
« Réécriture du
manuel concernant larrêté du 3 octobre 1995 » rapport
de stage F. PICOT session SPIBH 2000-2001
« Contribution à une réflexion
sur le contrôle qualité en mammographie » rapport de
B. BILLON C. VERALDO session SPIBH 2002-2003
« Réalisation dun outil
danalyse prévisionnelle des coûts de maintenance liés
à lapplication des règlements en vigueur » rapport
de G . GERMAIN session DESS TBH 2001-2002
« Processus de mise en place et
d'évolution du guide des bonnes pratiques biomédicales en
établissements de santé » rapport de M. Dhorne,P. Tappie
session DESS TBH 2002-2003
Etat de l'art des missions des services
biomédicaux, réflexions sur des bonnes pratiques de l'ingénierie
biomédicale, G. MANIBAL - C. RONCALLI , session DESS TBH 2000-2001
Création d'un service biomédical
au Centre Hospitalier de Valence, Y. Rochais, Stage DESS TBH 1999
Sites Internet
- http://www.anaes.fr
- http://www.legifrance.gouv.fr
- http://utc.fr/~farges
- http://www.afssaps.sante.fr
- http://www.afnor.fr
Annexes
Annexe 1 : méthodologie
de lélaboration dune enquête :
L'élaboration d'une enquête
pertinente doit être le fruit d'une réflexion afin de ne pas
recueillir des informations inappropriées ou inutiles aboutissant
à une mauvaise analyse et interprétation. A cet effet, il
convient de respecter une méthodologie. La littérature en
propose de nombreuses, cependant, le principe de base reste toujours le
même.
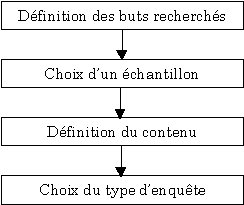
Les différentes étapes pour élaborer
une enquête
Définition des buts recherchés
: Il est indispensable de s'interroger sur les objectifs à atteindre.
Cela permettra à la fois de s'assurer de la pertinence de chaque
question mais aussi de structurer leur enchaînement.
Choix d'un échantillon : Un questionnaire
ne peut cibler qu'un type de client. Dans le contexte biomédical,
les clients sont les services cliniques et le service économique
Le choix du client doit être associé aux buts recherchés.
Ensuite, il est important de déterminer la ou les personnes, parmi
la catégorie de clients, qui sera sondée. En effet, le service
biomédical dispose de plusieurs interlocuteurs au sein d'un même
service clinique.
Définition du contenu : Le contenu
du questionnaire doit traduire l'objectif de la recherche, en d'autres
termes, la question posée en fonction du but donné, doit
susciter une réponse en relation avec le but poursuivi et traduisant
fidèlement l'attitude de l'enquêté.
L'objectif de l'enquête étant
fixé, il suffit de préciser les thèmes à aborder.
Les questions doivent être simples
pour éviter l'incompréhension et au contraire susciter les
réponses vraies. Il faut distinguer les questions fermées
(à choix unique ou à choix multiple) des questions ouvertes
(numériques ou non numériques).
La technique dite de l'entonnoir, c'est
à dire une enquête commençant par des questions générales
et simples pour aborder progressivement des questions de plus en plus précises,
doit être appliquée.
Choix du type de l'enquête: L'enquête
peut être à administration :
- directe :
le client répond lui-même au questionnaire, sans la présence
d'un enquêteur.
- indirecte :
c'est une interview, l'enquêteur complète lui-même le
questionnaire à partir des réponses fournies par le client.
Dans le cas d'une enquête à
administration indirecte, il est nécessaire de réfléchir
sur le choix de l'enquêteur :
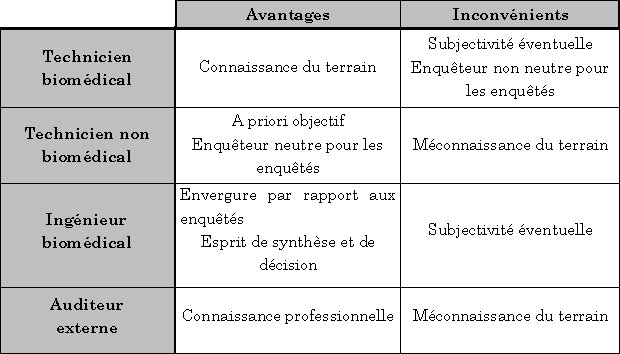
Choix de l'enquêteur pour l'enquête
de satisfaction client
Règles d'interprétation des résultats
d'une enquête :
Il n'existe pas de méthodologie «
type » permettant l'interprétation des résultats d'une enquête,
cependant quelques règles doivent être respectées afin de
mener, au plus juste, ce travail :
Analyser les réponses
par type de questions.
Prioriser les remarques manuscrites
: mettre en évidence les points négatifs.
Outre le cliché initial donné
par l'enquête, l'évaluation portera surtout sur l'évolution
de la satisfaction des clients. Il serait donc plus intéressant,
pour une démarche plus pertinente, de cibler les enquêtés
sur les points ayant posé problème.
Avant de conclure sur une enquête,
vérifier que :
- l'enquête
a été scrupuleusement réalisée
- qu'elle apporte
bien une réponse à la problématique de départ
- les résultats
sont fiables
- leur analyse
a été méthodiquement effectuée
- les destinataires
des conclusions sont identifiés
- le contenu
des conclusions correspond à son destinataire
Cibler les axes prioritaires d'action, apporter
dans un premier temps une réponse aux insatisfactions les plus flagrantes.
Informer les clients des résultats.
Annexe 2 : enquête

|
OUI
|
NON
|
|
1. Tenez-vous directement un inventaire des dispositifs médicaux
concernés ?
|
|
|
|
2. Avez-vous un listing des dispositifs médicaux soumis :
à lobligation de maintenance ?à lobligation de contrôle
qualité ?
|
|
|
|
3.Tenez-vous à jour un registre de maintenance et de contrôle
qualité ?
si oui, sous quel forme (classeur, fichier informatique
)
|
|
|
|
4.Avez-vous mis en uvre les nouvelles modalités de contrôle
qualité des mammographes?
(Mettre NC si non concerné)
|
|
|
|
5.Bénéficiez-vous dun logiciel de GMAO ?Si oui, larrêté
entraîne-til une modification importante de
celui ci ?
|
|
|
|
6.Possédez-vous des moyens nécessaires aux contrôles
qualités des équipements de radiologie
(fantôme, testeurs
) ?
|
|
|
|
7.Comptez-vous en acquérir le cas échéant :en achat
?en location ?
prêt inter-établissement ?
|
|
|
|
8. Avez-vous formé les différents acteurs aux contrôles
qualités(utilisateurs, techniciens
biomédicaux
) ?
|
|
|
|
9.Les contrôles qualités internes sont réalisés
par :
les utilisateurs, les biomédicaux ou les prestataires ?
|
|
|
10. Selon vous, est-il opportun dexternaliser :
la maintenance du matériel de radiologie?
le contrôle qualité du matériel de radiologie?
|
|
|
|
11. Serait-il judicieux dévaluer ces prestations de contrôle
lors de lachat des DM ?
|
|
|
|
12. Avez-vous mis en place un groupe de travail suite à larrêté
?
(chef de service, personnel biomédical, manipulateur, administratif
)
|
|
|
|
13. Votre budget actuel peut-il subvenir aux obligations de cet arrêté
?
|
|
|
|
14. Avez-vous pu présenter une politique de maintenance à
vos instances ?
|
|
|
|
15. Un programme dassurance qualité devient -il une nécessité
?
(ISO, Guide des Bonnes Pratiques Biomédicales
)
|
|
|
|
16.Rencontrez-vous des difficultés à mettre en application
cet arrêté ?
Si oui, pourquoi ? (une note annexe peut-être jointe)
|
|
|
|
17.Au regard de cette nouvelle réglementation, quelles évolutions
voyez-vous au sein de votre service
biomédical ? (classer par ordre de priorités de 1à
4) plus de techniciens
plus de formations
plus dECME
plus de budgets
autre (précisez)
|
|
Annexe 3 : Décret
du 5 décembre 2001
J.O n° 284 du 7 décembre 2001
page 19481
Textes généraux
Ministère de l'emploi et de la
solidarité
Décret no 2001-1154 du 5 décembre
2001 relatif à l'obligation de maintenance et au contrôle
de qualité des dispositifs médicaux prévus à
l'article L. 5212-1 du code de la santé publique (troisième
partie : Décrets)
NOR: MESP0123968D
Le Premier ministre,
Sur le rapport de la ministre de l'emploi
et de la solidarité et du ministre délégué
à la santé,
Vu la directive 97/43/EURATOM du Conseil
du 30 juin 1997 relative à la protection sanitaire des personnes
contre les dangers des rayonnements inonisants lors de l'exposition à
des fins médicales, remplaçant la directive 84/466/EURATOM
;
Vu le code de la santé publique,
notamment ses articles L. 5212-1, L. 5212-2, L. 5312-1, L. 6122-11, L.
6122-13, R. 665-7, R. 665-12 et R. 665-49,
Décrète :
Art. 1er. - I. - Il est inséré,
à la section 1 du chapitre Ier du livre V bis du code de la santé
publique (troisième partie : Décrets), les articles D. 665-5-1
à D. 665-5-4 ainsi rédigés :
« Art. D. 665-5-1. - Pour l'application
des dispositions du présent livre :
« 1o On entend par "exploitant"
d'un dispositif médical toute personne physique ou morale assurant
la responsabilité juridique de l'activité requérant
l'utilisation de ce dispositif ;
« 2o On entend par "maintenance"
d'un dispositif médical l'ensemble des activités destinées
à maintenir ou à rétablir un dispositif médical
dans un état ou dans des conditions données de sûreté
de fonctionnement pour accomplir une fonction requise ; les conditions
de réalisation de la maintenance sont fixées contractuellement,
s'il y a lieu, entre le fabricant ou le fournisseur de tierce maintenance
et l'exploitant ;
« 3o On entend par "contrôle
de qualité" d'un dispositif médical l'ensemble des opérations
destinées à évaluer le maintien des performances revendiquées
par le fabricant ou, le cas échéant, fixées par le
directeur général de l'Agence française de sécurité
sanitaire des produits de santé ; le contrôle de qualité
est dit interne, s'il est réalisé par l'exploitant ou sous
sa responsabilité par un prestataire ; il est dit externe, s'il
est réalisé par un organisme indépendant de l'exploitant,
du fabricant et de celui qui assure la maintenance du dispositif.
« Art. D. 665-5-2. - L'exploitant
veille à la mise en oeuvre de la maintenance et des contrôles
de qualité prévus pour les dispositifs médicaux qu'il
exploite. La maintenance est réalisée soit par le fabricant
ou sous sa responsabilité, soit par un fournisseur de tierce maintenance,
soit par l'exploitant lui-même.
« Art. D. 665-5-3. - En application
de l'article L. 5212-1, le ministre chargé de la santé après
avis du directeur général de l'Agence française de
sécurité sanitaire des produits de santé arrête
la liste des dispositifs médicaux soumis à l'obligation de
maintenance, celle des dispositifs médicaux soumis au contrôle
de qualité interne et la liste des dispositifs médicaux soumis
au contrôle de qualité externe.
« Art. D. 665-5-4. - Pour chacun
des dispositifs soumis au contrôle de qualité interne ou externe,
le directeur général de l'Agence française de sécurité
sanitaire des produits de santé définit les modalités
particulières de ce contrôle, en fonction des dispositifs,
par décision publiée au Journal officiel de la République
française. Pour ce qui concerne les dispositifs médicaux
exposant les personnes à des rayonnements ionisants, les décisions
du directeur général de l'Agence française de sécurité
sanitaire des produits de santé sont prises au vu de l'avis de l'organisme
désigné par l'autorité administrative compétente
en matière de radioprotection.
« Le directeur général
de l'Agence française de sécurité sanitaire des produits
de santé fixe notamment :
« - les critères d'acceptabilité
auxquels doivent répondre les performances ou les caractéristiques
des dispositifs médicaux soumis au contrôle de qualité
interne ou externe ;
« - la nature des opérations
de contrôle à mettre en oeuvre pour s'assurer du maintien
des performances des dispositifs médicaux et les modalités
de leur réalisation ;
« - la périodicité
des contrôles et les situations nécessitant un contrôle
en dehors des contrôles périodiques ;
« - la nature des opérations
de maintenance des dispositifs médicaux qui nécessitent un
nouveau contrôle en dehors des contrôles périodiques
;
« - les recommandations en matière
d'utilisation et de remise en conformité compte tenu des dégradations
ou des insuffisances de performances ou des caractéristiques constatées
ainsi que, le cas échéant, les délais laissés
à l'exploitant pour remettre en conformité les dispositifs.
»
II. - Il est inséré, au
chapitre Ier du livre V bis du code de la santé publique (troisième
partie : Décrets) une section 10 ainsi rédigée :
« Section 10
« Mise en oeuvre de l'obligation
de maintenance
et du contrôle de qualité
« Art. D. 665-5-5. - Pour les dispositifs
médicaux mentionnés à l'article D. 665-5-3, l'exploitant
est tenu :
« 1o De disposer d'un inventaire
des dispositifs qu'il exploite, tenu régulièrement à
jour, mentionnant pour chacun d'eux les dénominations commune et
commerciale du dispositif, le nom de son fabricant et celui du fournisseur,
le numéro de série du dispositif, sa localisation et la date
de sa première mise en service ;
« 2o De définir et mettre
en oeuvre une organisation destinée à s'assurer de l'exécution
de la maintenance et du contrôle de qualité interne ou externe
des dispositifs dont il précise les modalités, qui sont transcrites
dans un document ; dans les établissements de santé, cette
organisation est adoptée après avis des instances médicales
consultatives ; elle est portée à la connaissance des utilisateurs
; les changements de cette organisation donnent lieu, sans délai,
à la mise à jour du document ;
« 3o De disposer d'informations
permettant d'apprécier les dispositions adoptées pour l'organisation
de la maintenance et du contrôle de qualité interne ou externe,
ainsi que les modalités de leur exécution ;
« 4o De mettre en oeuvre les contrôles
prévus par l'article D. 665-5-4 ;
« 5o De tenir à jour, pour
chaque dispositif médical, un registre dans lequel sont consignées
toutes les opérations de maintenance et de contrôle de qualité
interne ou externe, avec pour chacune d'elles l'identité de la personne
qui les a réalisées et, le cas échéant, de
son employeur, la date de réalisation des opérations effectuées
et, le cas échéant, la date d'arrêt et de reprise d'exploitation
en cas de non-conformité, la nature de ces opérations, le
niveau de performances obtenu, et le résultat concernant la conformité
du dispositif médical ; ce registre doit être conservé
cinq ans après la fin d'exploitation du dispositif,
sauf dispositions particulières
fixées par décision du directeur général de
l'Agence française de sécurité sanitaire des produits
de santé pour certaines catégories de dispositifs ;
« 6o De permettre l'accès
aux dispositifs médicaux et aux informations prévues par
le présent article à toute personne en charge des opérations
de maintenance et de contrôle de qualité.
« Art. D. 665-5-6. - Le contrôle
de qualité externe des dispositifs médicaux est réalisé
par des organismes agréés à cet effet par décision
du directeur général de l'Agence française de sécurité
sanitaire des produits de santé publiée au Journal officiel
de la République française. L'agrément précise
les tâches pour lesquelles il est accordé.
« L'agrément est accordé
pour une durée de cinq ans renouvelable, en fonction des garanties
d'indépendance et de compétence présentées,
de l'expérience acquise dans le domaine considéré
et des moyens dont l'organisme dispose pour exécuter les tâches
pour lesquelles il est agréé. La composition du dossier de
demande d'agrément est fixée par arrêté du ministre
chargé de la santé après avis du directeur général
de l'Agence française de sécurité sanitaire des produits
de santé.
« Les organismes qui satisfont aux
normes les concernant, transposant les normes européennes harmonisées,
dont les références ont été publiées
au Journal officiel de la République française, sont présumés
répondre à ces critères.
« Les organismes doivent s'engager
à permettre aux personnes désignées par le directeur
général de l'Agence française de sécurité
sanitaire des produits de santé d'accéder à leurs
locaux et de procéder à toute investigation, afin de vérifier
qu'ils continuent de satisfaire aux conditions de l'agrément.
« Les organismes s'engagent en outre
à communiquer au directeur général de l'Agence française
de sécurité sanitaire des produits de santé toute
modification des conditions d'exercice de leurs activités, telles
qu'elles sont énoncées dans leur demande d'agrément.
« Si un organisme agréé
cesse de remplir les conditions qui ont permis son agrément, celui-ci
peut être retiré par décision du directeur général
de l'Agence française de sécurité sanitaire des produits
de santé après que le responsable de l'organisme a été
mis à même de présenter ses observations.
« Les organismes agréés
établissent un rapport annuel d'activité qu'ils communiquent
au directeur général de l'Agence française de sécurité
sanitaire des produits de santé. Ce rapport d'activité mentionne,
d'une part, la part du chiffre d'affaires relative aux contrôles
effectués dans le cadre de cet agrément et, d'autre part,
pour chacun des contrôles de qualité effectués, le
nom de l'exploitant, le dispositif contrôlé, la nature des
contrôles réalisés et les non-conformités observées.
« Art. D. 665-5-7. - Les organismes
agréés mettent en oeuvre, à la demande de l'exploitant,
les contrôles conformément aux dispositions particulières
prévues à l'article D. 665-5-4.
« Chaque contrôle de qualité
externe donne lieu à l'établissement d'un rapport relatif
au maintien des performances du dispositif contrôlé. Ce rapport
mentionne le nom de l'exploitant, le dispositif contrôlé,
la nature des contrôles effectués et les non-conformités
observées. Il est remis à l'exploitant qui en consigne un
exemplaire dans le registre mentionné au 5o de l'article D. 665-5-5.
« Art. D. 665-5-8. - Dans le cas
où un contrôle de qualité met en évidence une
dégradation des performances ou des caractéristiques du dispositif,
l'exploitant prend des mesures appropriées relatives à l'utilisation
et procède à la remise en conformité du dispositif
conformément aux dispositions prévues à l'article
D. 665-5-4.
« Si les dégradations des
performances constatées sont susceptibles d'entraîner un risque
d'incident tel que prévu à l'article L. 5212-2, celui-ci
fait l'objet d'un signalement en application du même article, accompagné
du rapport mentionné à l'article D. 665-5-7, si le dispositif
a fait l'objet d'un contrôle de qualité externe.
« Art. D. 665-5-9. - Dans le cas
du contrôle de qualité externe, la remise en conformité
des dispositifs est attestée par les résultats conformes
d'un second contrôle de qualité réalisé sur
le dispositif selon les dispositions prévues à l'article
D. 665-5-7.
« Si, après ce second contrôle,
les performances attendues du dispositif ne sont toujours pas atteintes,
l'organisme agréé informe le directeur général
de l'Agence française de sécurité sanitaire des produits
de santé.
« Art. D. 665-5-10. - Dans le cas
où le contrôle de qualité a conduit au signalement
d'un risque d'incident prévu par l'article D. 665-5-8, l'exploitant
notifie au directeur général de l'Agence française
de sécurité sanitaire des produits de santé la remise
en conformité du dispositif médical ou sa mise hors service
définitive.
« En cas de remise en conformité,
si le dispositif médical a fait l'objet d'un contrôle de qualité
externe, l'exploitant communique à l'Agence française de
sécurité sanitaire des produits de santé le rapport
mentionné à l'article D. 665-5-7 relatif au second contrôle.
« Art. D. 665-5-11. - Quand le directeur
général de l'Agence française de sécurité
sanitaire des produits de santé a connaissance qu'un exploitant
d'un dispositif médical soumis au contrôle de qualité
n'a pas mis en oeuvre les dispositions prévues par le présent
code, il met en demeure l'exploitant de faire procéder à
ce contrôle.
« Art. D. 665-5-12. - En cas de
non-conformité d'un dispositif médical, constatée
à la suite d'un contrôle de qualité, le directeur général
de l'Agence française de sécurité sanitaire des produits
de santé informe le préfet de région, le directeur
de la caisse régionale d'assurance maladie, et, le cas échéant,
le directeur de l'agence régionale d'hospitalisation. »
Art. 2. - La ministre de l'emploi et de
la solidarité et le ministre délégué à
la santé sont chargés, chacun en ce qui le concerne, de l'exécution
du présent décret, qui sera publié au Journal officiel
de la République française.
Fait à Paris, le 5 décembre
2001.
Lionel Jospin
Par le Premier ministre :
La ministre de l'emploi et de la solidarité,
Elisabeth Guigou
Le ministre délégué
à la santé,
Bernard Kouchner
Annexe 4 : Arrêté
du 3 mars 2003
J.O n° 66 du 19 mars 2003 page 4848
Décrets, arrêtés,
circulaires
Textes généraux
Ministère de la santé, de
la famille et des personnes handicapées
Arrêté du 3 mars 2003 fixant
les listes des dispositifs médicaux soumis à l'obligation
de maintenance et au contrôle de qualité mentionnés
aux articles L. 5212-1 et D. 665-5-3 du code de la santé publique
NOR: SANP0320928A
Le ministre de la santé, de la famille
et des personnes handicapées,
Vu la directive 97/43/EURATOM du Conseil
du 30 juin 1997 relative à la protection sanitaire des personnes
contre le danger des rayonnements ionisants lors des expositions à
des fins médicales remplaçant la directive 84/466/EURATOM
;
Vu le code de la santé publique,
notamment les articles L. 5212-1 et D. 665-5-3 ;
Vu l'avis du directeur général
de l'Agence française de sécurité sanitaire des produits
de santé,
Arrête :
Article 1
En application de l'article D. 665-5-3
du code de la santé publique, sont fixées :
- à l'annexe I du présent
arrêté, la liste des dispositifs médicaux soumis à
l'obligation de maintenance ;
- à l'annexe II du présent
arrêté, la liste des dispositifs médicaux soumis au
contrôle de qualité interne ;
- à l'annexe III du présent
arrêté, la liste des dispositifs médicaux soumis au
contrôle de qualité externe.
Article 2
L'obligation de maintenance à laquelle
sont tenus les exploitants en application de l'article D. 665-5-3 du code
de la santé publique doit être effective au plus tard le 1er
janvier 2004 pour les dispositifs médicaux énumérés
aux paragraphes 1 à 4 de l'annexe I, à l'exception toutefois
des dispositifs destinés à la mammographie ou à l'ostéodensitométrie
pour lesquels cette obligation entre en vigueur immédiatement.
Pour les dispositifs médicaux énumérés
au paragraphe 5 de l'annexe I, cette obligation de maintenance doit être
effective au plus tard le 1er janvier 2004 lorsqu'ils sont mis en service
postérieurement à la date de publication du présent
arrêté et au plus tard le 1er janvier 2005 pour ceux déjà
mis en service à la date de la publication du présent arrêté.
Article 3
Le directeur général de l'Agence
française de sécurité sanitaire des produits de santé
est chargé de l'exécution du présent arrêté,
qui sera publié au Journal officiel de la République française.
Fait à Paris, le 3 mars 2003.
Pour le ministre et par délégation
:
Le directeur général de la
santé,
L. Abenhaïm
A N N E X E I
LISTE DES DISPOSITIFS MÉDICAUX SOUMIS
À L'OBLIGATION DE MAINTENANCE
A l'exception des dispositifs médicaux
pour lesquels ils sont en mesure de justifier qu'une maintenance est inutile
en raison de leur conception ou de leur destination, les exploitants sont
tenus de s'assurer de la maintenance des dispositifs médicaux suivants
:
1-1. Dispositifs médicaux nécessaires
à la production et à l'interprétation des images de
radiodiagnostic ;
1-2. Dispositifs médicaux nécessaires
à la définition, à la planification et à la
délivrance des traitements de radiothérapie ;
1-3. Dispositifs médicaux nécessaires
à la réalisation des actes de médecine nucléaire
;
1-4. Dispositifs médicaux à
finalité diagnostique ou thérapeutique exposant les personnes
à des rayonnements ionisants en dehors des dispositifs médicaux
mentionnés aux paragraphes 1-1, 1-2 et 1-3 ;
1-5. Dispositifs médicaux des classe
IIb et III résultant des règles de classification prévues
à l'annexe IX du livre V bis du code de la santé publique
( deuxième partie : Décrets en Conseil d'Etat), autres que
les dispositifs mentionnés aux paragraphes 1-1, 1-2, 1-3 et 1-4.
A N N E X E I I
LISTE DES DISPOSITIFS MÉDICAUX SOUMIS
AU CONTRÔLE DE QUALITÉ INTERNE
2-1. Dispositifs médicaux nécessaires
à la production et à l'interprétation des images de
radiodiagnostic ;
2-2. Dispositifs médicaux nécessaires
à la définition, à la planification et à la
délivrance des traitements de radiothérapie ;
2-3. Dispositifs médicaux nécessaires
à la réalisation des actes de médecine nucléaire
;
2-4. Dispositifs médicaux à
finalité diagnostique ou thérapeutique exposant les personnes
à des rayonnements ionisants autres que les dispositifs médicaux
mentionnés aux paragraphes 2-1, 2-2 et 2-3.
A N N E X E I I I
LISTE DES DISPOSITIFS MÉDICAUX SOUMIS
AU CONTRÔLE DE QUALITÉ EXTERNE
3-1. Dispositifs médicaux nécessaires
à la production et à l'interprétation des images de
radiodiagnostic ;
3-2. Dispositifs médicaux nécessaires
à la définition, à la planification et à la
délivrance des traitements de radiothérapie ;
3-3. Dispositifs médicaux nécessaires
à la réalisation des actes de médecine nucléaire
;
3-4. Dispositifs médicaux à
finalité diagnostique ou thérapeutique exposant les personnes
à des rayonnements ionisants autres que les dispositifs médicaux
mentionnés aux paragraphes 3-1, 3-2 et 3-3.