|
Avertissement
|
Si vous arrivez
directement sur cette page, sachez que ce travail est un rapport
d'étudiants et doit être pris comme tel. Il peut donc
comporter des imperfections ou des imprécisions que le lecteur
doit admettre et donc supporter. Il a été
réalisé pendant la période de formation et
constitue avant-tout un travail de compilation bibliographique,
d'initiation et d'analyse sur des thématiques associées
aux concepts, méthodes, outils et expériences sur les
démarches qualité dans les organisations. Nous ne faisons aucun usage commercial et la
duplication est libre. Si vous avez des raisons de contester ce droit
d'usage, merci de nous en faire part .
L'objectif de la présentation sur le Web est de
permettre l'accès à l'information et d'augmenter ainsi
les échanges professionnels. En cas d'usage du document,
n'oubliez pas de le citer comme source bibliographique. Bonne
lecture...
|
LA MISE EN PLACE DE LA
CERTIFICATION ISO 13485
AU SEIN D’AXESS VISION TECHNOLOGY
|

Anne
HERMELIN
|
|
|
RESUME
Axess
Vision Technology (AVT) est une Start-Up œuvrant dans le domaine du
biomédical.
Afin
d’introduire son dispositif, un endoscope à usage unique, sur le
marché début
2010, AVT a la volonté de mettre en place un système de
management de la
qualité répondant à la norme ISO 13485. La
certification est prévue pour
décembre 2009.
La mission du stage
présenté dans ce
rapport est de mettre en place ce système qualité. Pour
cela, il faut
identifier les processus de la société, rédiger
l’ensemble des documents
qualité répondant aux exigences de la norme et
sensibiliser l’ensemble des acteurs
de la société au fonctionnement du système
qualité.
Mots clés : Système
qualité, ISO 13485,
dispositif médical
|
ABSTRACT
Axess
Vision Technology (AVT) is a Start-up working in the biomedical field.
To
introduce its device, a single-use endoscope, on the market at the
beginning of
2010, AVT wants to set up a quality management system meeting the
standard ISO
13485. The certification is planned for December, 2009.
The
mission of the training course presented in this report is to implement
this
quality system. For that purpose, it is necessary to identify the
processes of
the company, to draft all the quality documents answering the standard
requirements and to make sensitive all the actors of the company in the
functioning of the quality system.
Key words : Quality
system, ISO 13485, medical device
|
Remerciements
Je
tiens à remercier dans un premier temps, toute l’équipe
pédagogique de l’UTC et les intervenants professionnels
responsables de la formation de
management de la qualité, pour avoir assuré
la partie
théorique de celle-ci.
Je
remercie également Monsieur Farges pour l’aide et les conseils
concernant les
missions évoquées dans ce rapport, qu’il m’a
apporté lors de sa visite dans
l’entreprise.
Je
tiens à remercier tout particulièrement et à
témoigner toute ma reconnaissance
aux personnes suivantes, pour l’expérience enrichissante et
pleine d’intérêt
qu’elles m’ont fait vivre durant ces cinq mois au sein de l’entreprise Axess
Vision Technology:
Monsieur
Fructus, PDG de la société Axess Vision Technology, mon
tuteur, pour son
accueil et la confiance qu’il m’a accordés dès mon
arrivée dans l’entreprise.
Monsieur
Mathieu, Directeur Technique de la société Axess Vision
Technology, pour le
temps qu’il m’a consacré tout au long de cette période
afin répondre à
l’ensemble de mes questions.
Mademoiselle
Hasnae Abou pour son accueil chaleureux ; sa disponibilité
et sa patience
pour m’intégrer dans le projet.
Madame
Justel, Madame Labbé et l’ensemble du personnel d’Axess Vision
Technology, pour
leur coopération professionnelle tout au long de ces cinq mois.
Sommaire
Remerciements. 2
Table des
illustrations. 5
Introduction.. 6
Chapitre 1 : Contexte, enjeux et
problématique. 7
1- Présentation
d’Axess Vision
Technology. 7
1.1
Objectif d’Axess Vision Technology. 7
1.2
Historique de la société. 7
1.3
Organisation de la société Axess Vision Technology. 7
2- Contexte. 10
2.1
Concurrence : entreprises et produits. 10
3- Présentation
de la technologie. 11
3.1 La
technologie utilisée actuellement 11
3.2 Les contraintes de la
technologie
actuellement utilisée. 12
3.2.1 Un
équipement non stérile. 12
3.2.2 Les conséquences de
l’endoscopie
traditionnelle. 13
3.3 Les
solutions d’Axess Vision Technology. 15
3.3.1
Historique du dispositif novateur : le DispoFlex. 15
3.3.2
Technologie. 16
3.3.3 Les
avantages de cette nouvelle technologie. 17
Chapitre 2 : Approche qualité
développée. 18
1- Etat
des lieux de la mission.. 18
2-
Clarification de la problématique. 19
3-
Objectifs et enjeux. 20
4-
Méthodologie de travail 21
5-
Planification de différentes actions à accomplir 23
6-
Analyse de risques. 24
Chapitre 3 : Résultats, discussion et
perspectives. 25
1-
Résultats. 25
1.1
La Cartographie de processus. 25
1.2
La politique Qualité et la lettre de cadrage. 27
1.3
Les procédures. 29
1.4
Le Tableau de Bord. 30
1.5
Le Manuel Qualité. 31
2- Discussion
et perspectives. 31
Conclusion.. 32
Bilan de la mission.. 32
Apport à l’entreprise. 32
Apport personnel 32
Abbreviations. 34
Glossaire. 35
Bibliographie. 36
Les
référentiels normatifs. 36
Ouvrage
interne. 36
Les
sites consultés. 36
Annexes. i
ANNEXE 1 : Liste des lois
pour la
définition des spécifications
réglementaires. i
ANNEXE
2 : PR01 : GESTION DOCUMENTAIRE.. ii
ANNEXE 3 : SOMMAIRE
DU
MANUEL QUALITE
.. ix
Figure
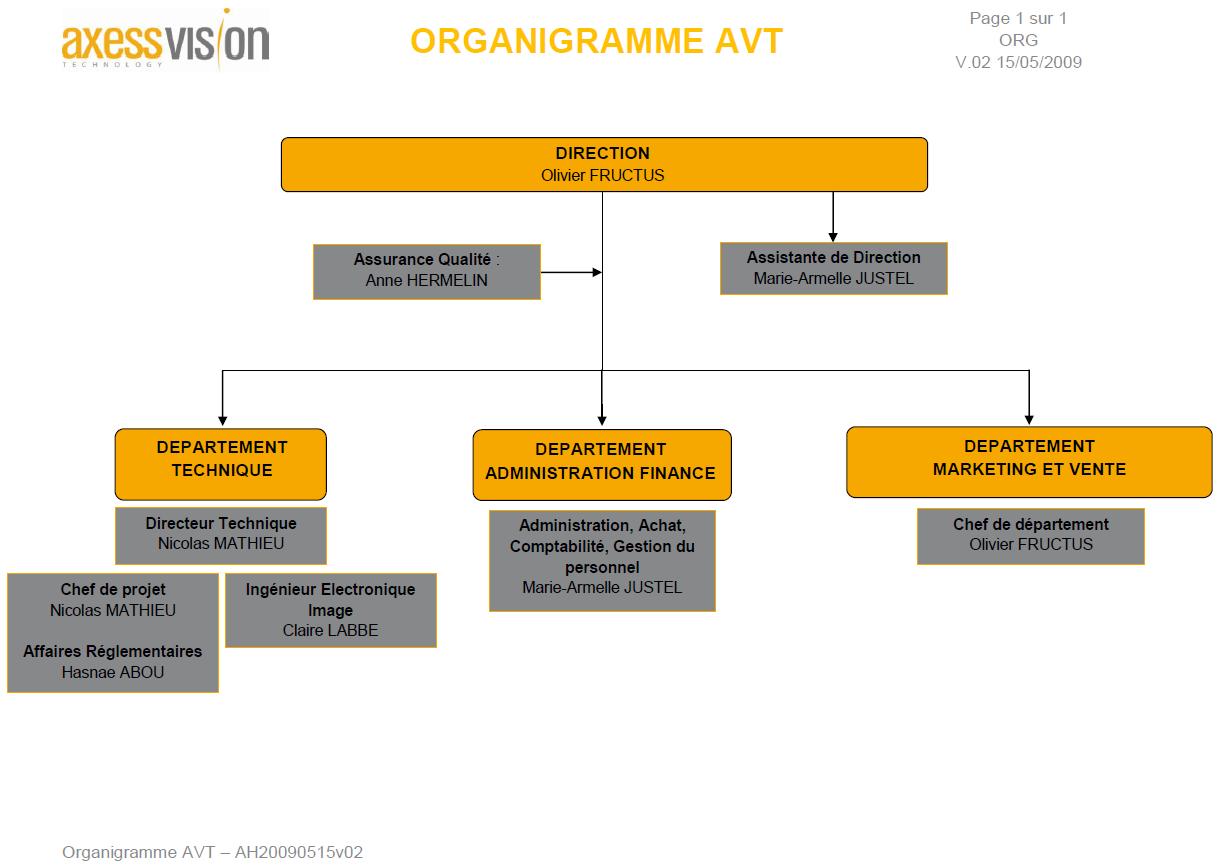
1 Organigramme de la société Axess Vision Technology. 9
Figure
2 Part du marché des fabricants d'endoscope (%) 10
Figure
3 Endoscope flexible Traditionnel 11
Figure
4 "Tour" avec endoscope flexible. 12
Figure
5 Détérioration fréquemment découverte sur
des endoscopes traditionnels. 13
Figure
6 Processus choisi par Axess Vision Technology. 15
Figure
7 DispoFlex, Schéma global 16
Figure
8 Ensemble des éléments de l'endoscope. 17
Figure
9 Voie de passage d'un bronchoscope. 17
Figure
10 QQOQCP.. 19
Figure
11 Planification Dynamique Stratégique. 20
Figure
12 Rétro planning du déroulement du stage. 23
Figure
13 Diagramme de décisions. 24
Figure
14 Cartographie des processus. 26
Figure
15 Politique Qualité. 27
Figure
16 Lettre de Cadrage. 28
Tableau
1 Procédures exigées par la norme ISO 13485. 29
Tableau
2 Tableau de bord. 30
Tableau
3 Bilan des missions réalisées
. 32
La Qualité est
l’aptitude d'un ensemble de caractéristiques intrinsèques
à satisfaire des
exigences. (ISO 9000 2000).[1]
Elle est devenue incontournable ; ses enjeux
sont
multiples :
·
En termes d’image :
Elle concourt à
la renommée, à la réputation,
à l’image de marque et à l’assurance qualité
accordée par les clients
·
En termes de
compétitivité :
La
compétitivité des prix est liée
aux surcoûts de la non qualité. La qualité permet
également une compétitivité en
termes de délai, d’image, de progrès technique et
d’innovation.
·
En termes de cohésion sociale
dans l’entreprise
:
La qualité permet
une autonomie plus
forte du personnel, une rigueur dans le travail, l’initiative et la
responsabilisation
de chacun.
Axess Vision Technology (AVT) est une jeune
entreprise
innovante qui œuvre dans le domaine de l’équipement
Médical. Elle a pour
objectif d’obtenir la certification ISO 13485 d’ici fin 2009.[2] Ce
rapport,
réalisé dans le cadre de mon stage de fin d’études
de Master 2 Management de la
Qualité à l’Université de Technologie de
Compiègne (UTC), a pour principal objectif
de présenter la démarche entreprise pour mettre en place
le système qualité
répondant à la norme ISO 13485 au sein d’AVT.
Lors de ce stage,
j’ai également
travaillé en collaboration avec le service des affaires
réglementaires. Ceci
m’a permis de travailler sur des normes spécifiques au domaine
biomédical autres
que la norme ISO 13485. En effet ; les spécifications
réglementaires,
définies par le département Affaires
Réglementaires, émanent de l’ISO 62 366,
DM-Application de l’ingénierie de l’aptitude aux dispositifs
médicaux. [3]
Les spécifications réglementaires,
définies par le
département Affaires Réglementaires, représentent
les exigences des différentes
lois de pays où nous envisageons de commercialiser le produit.
(Cf Annexe1 :
Liste des lois pour la définition des spécifications
réglementaires).
Ce rapport se concentrera plus
particulièrement sur la
mise en place du système qualité répondant
à la norme ISO 13485.
1-
Présentation
d’Axess Vision Technology
Axess Vision Technology est une
société innovante créé en
2006. Elle est spécialisée dans le développement,
la fabrication et la vente de
vidéo-endoscopes souples, stériles à usage unique.
La société Axess Vision
Technology se situe 5 place Jean Jaurès à Tours.
1.1 Objectif d’Axess Vision
Technology
L’objectif de l’entreprise est de mettre sur le
marché une
nouvelle technologie, un vidéo-endoscope souple à usage
unique en 2010.
Pour cela, l’entreprise se concentre pour un
premier temps
sur le marché interne européen, le label CE permettant la
mise sur le marché du
produit dans tous les pays de l’Union Européenne.
L’ensemble des sous-traitants avec lesquels AVT
travaille sont
certifiés par les Food and Drug Administration (FDA),
l’administration
américaine des denrées alimentaires et des
médicaments, qui a, entre autres, le
mandat d'autoriser la commercialisation des médicaments / des
instruments
médicaux sur le territoire des États-Unis. Dans un second
temps AVT travaille
pour l’obtention d’une homologation US afin de commercialiser son
dispositif sur
le territoire nord américain. [5]
Il est donc crucial qu’elle obtienne la
certification ISO
13485, norme qui précise les exigences des systèmes de
management de la qualité
(SMQ) pour l'industrie des dispositifs médicaux. L’audit blanc
est prévu en
décembre 2009.
1.2 Historique de la
société
La société Axess Vision Technology a
été créée au début de
l’année 2006 sous forme d’une société anonyme et
avec son siège social
initialement à Lyon.
1.3 Organisation de la
société Axess
Vision Technology
La
société est une
« start-up» composée de quatre cadres :
·
Olivier
Fructus : Président Directeur Général
·
Nicolas
Mathieu : Directeur Technique
·
Marie
Armelle Justel : Office Manager
·
Claire
Labbé : Ingénieur Électronique Image
retour sommaire
Axess Vision Technology a recruté des
stagiaires dans
plusieurs domaines :
·
Anne
Hermelin : Stagiaire Responsable Qualité
·
Hasnae
Abou : Stagiaire en Affaire Réglementaire
·
Alban
Psaila : stagiaire Optique
·
Samira
Benmerzoug : Stagiaire en Ressources Humaines
·
Kévin
Toublanc : Stagiaire Electronique et Informatique
L’organisation générale de
l’entreprise est retrouvée dans
l’organigramme ci-dessous :
2-
Contexte
2.1
Concurrence : entreprises et produits
La concurrence d’Axess
Vision Technology provient des fabricants
d’endoscopes traditionnels, même si les fabricants concurrents ne
vendent pas
actuellement d’endoscope médical à usage unique pour tous
les domaines
d’application, de l'endoscopie traditionnelle de diagnostic aux
endoscopies
thérapeutiques.
Un grand nombre de nouvelles
sociétés qui travaillent sur des systèmes
d’endoscopes
jetables ou semi-jetables ont été identifiées.
·
Micro-Invasive
Technologies – USA, endoscopes
semi-rigides
·
STM - Invendo –
Allemagne, endoscopes flexibles et
jetables pour coloscopies, mettant en œuvre un mécanisme de
commande
sophistiqué à joystick
·
Dioptik-France,
fibroscopes à usage unique pour
ENT.
Les concurrents principaux sont les
fabricants d’endoscopes
traditionnels. Les 3 principaux acteurs de l’industrie des endoscopes
flexibles
sont les fabricants de caméras et de lentilles japonais
traditionnels :
Olympus, Pentax, Fujinon.
Les autres fabricants d’endoscopes
flexibles, Storz, Wolf, ACMI, jouent
un rôle mineur dans ce segment.[4]
Figure
2 Part du
marché des fabricants d'endoscope (%)
Le bénéfice global de
l'endoscopie flexible dans la médecine humaine
(endoscopes, « tours », accessoires, maintenance, à
l’exception du processus de
désinfection, ainsi que des équipements et
infrastructures qui y sont liés) est
estimé à 1,7 milliards d'euros par an, avec un AAGR de
1,7 %.
Les trois principaux fabricants
disposent de leur propre réseau de
distribution, avec des ventes directes et des services de maintenance.
L’incidence de la maintenance sur les coûts et la
disponibilité des équipements
est très élevée, chaque endoscope subissant 2
à 3 défaillances par an pour un
budget total de 3.000€ par appareil. Les temps d’arrêt varient de
3 à 6
semaines. La durée de service d’une colonne vidéo est de
10 ans, et celle d’un
endoscope de seulement 3 ans.
Depuis la fin des années
1980 et le remplacement des faisceaux de fibres
optiques par des caméras CCD, aucune innovation significative
n’a été
introduite dans l’endoscopie traditionnelle.
Les fabricants
ont seulement amélioré les techniques existantes,
notamment en augmentant la résolution
et en agrandissant les images à l’écran.
Récemment, des options coûteuses ont
été proposées, telles que la flexibilité
variable du tube d’insertion, un zoom
mécanique et l’élastographie à ultrasons
combinée.[4]
3-
Présentation de la
technologie
3.1 La
technologie utilisée actuellement
Le premier vrai fibroscope date de 1957. Les
endoscopes flexibles ont été utilisés dans les
interventions de routine depuis
1971.
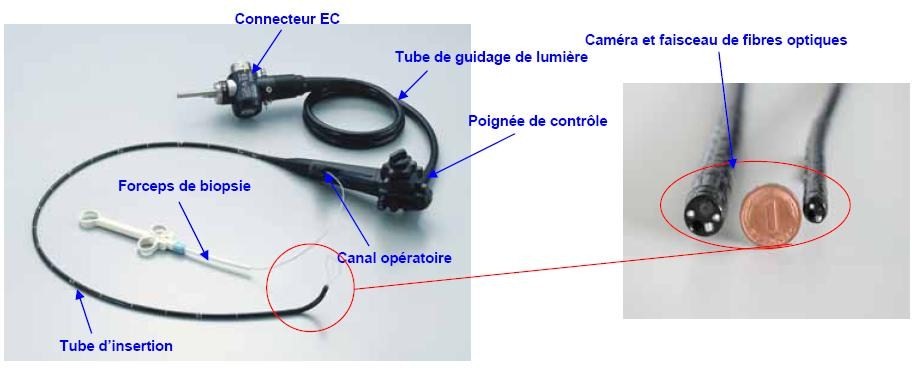
Figure
3
Endoscope flexible Traditionnel
Un endoscope flexible (diagnostique
et thérapeutique) est un dispositif
médical qui permet l’examen visuel de l’intérieur d’un
organe creux du corps.
Ce dispositif se compose d'un tube d'insertion introduit dans la
cavité par un
orifice naturel (bouche, anus, sinus, ...), qui véhicule de la
lumière et
renvoie au praticien une image par le biais d’une caméra ou d’un
faisceau de
fibres optiques placé à l’extrémité
distale. Le tube d’insertion peut également
disposer de canaux opérateurs destinés à des fins
de diagnostic pour aspirer,
rincer ou introduire des forceps de biopsie, ou à des fins
thérapeutiques avec
un instrument chirurgical quelconque (boucles, aiguilles, …).
Le tube d’insertion (la seule
partie de l’endoscope en contact avec le
patient) comprend une poignée de béquillage
(poignée de contrôle), permettant
au praticien de diriger le dispositif (direction, air, eau, image,
lumière).
L’extrémité distale du tube d’insertion peut être
recourbée à un angle de 180°.
L’extrémité proximale de l’endoscope appelée
« tube de guidage de lumière » est
reliée au module de base (la « tour d’endoscope »)
à des fins d’éclairage et de
traitement d’image.

Figure
4 "Tour"
avec endoscope flexible
Cet équipement volumineux et
coûteux n’est pas très maniable.
Afin d’insérer correctement
le tube d’insertion (par la bouche ou le
colon) tout en évitant de soumettre le patient à une
manipulation douloureuse
ou inconfortable, la plupart des examens sont réalisés
sous anesthésie de
courte durée, qui nécessite la présence d’un
anesthésiste (70 % des
explorations endoscopiques gastro-intestinales sont
réalisées sous
anesthésiques).
Les protocoles de
désinfection stricts récents ont également
contribué
au confinement de telles interventions dans les salles
d’opération ou des salles
spéciales destinées aux endoscopies situées au
sein du bloc opératoire. Rares
sont les endoscopies réalisées aujourd’hui au lit du
patient (c’est à dire en
soins intensifs et services d’urgence).
Le temps nécessaire à
la réalisation d’une endoscopie varie de 2 minutes
(gastroscopie) à 30 minutes pour une coloscopie
compliquée.[4]
3.2 Les
contraintes de la technologie actuellement utilisée
Les
endoscopes flexibles sont sensibles à la
température et ne peuvent pas être
stérilisés en autoclave, contrairement à
tous les autres équipements médicaux réutilisables
(c’est-à dire selon le
protocole français : stérilisation à la vapeur :
134 °C, pression de 2,5 bars
pendant une durée de 18 minutes).
Les
endoscopies traditionnelles ne sont
réalisées qu’avec des dispositifs qui doivent être
nettoyés et désinfectés
consciencieusement entre les examens, ainsi qu’à chaque
début et fin de
journée.
Pour un
examen de 5 minutes, ceci implique
un processus de décontamination double et une immersion de
jusqu’à 1 heure
Outre la
complexité du processus de désinfection en raison du
concept des endoscopes et
des protocoles de traitement sévères, l’intervention
humaine obligatoire
au cours du processus ne garantit ni une fiabilité à 100
%, ni la reproductibilité
des procédures de désinfection. Le démontage de
l’endoscope durant l’entretien
démontre clairement à quel point il est difficile de
prévenir l’invasion de
fluides à l’intérieur d’un endoscope réutilisable
et d’éviter la contamination
par du sang ou des protéines.[4]
Figure
5
Détérioration fréquemment
découverte sur
des endoscopes traditionnels
L’endoscopie telle qu’elle est
pratiquée jusqu’à présent présente des
risques
et des conséquences significatifs pour le patient, le praticien,
les centres
hospitaliers et la société.
Pour le patient :
·
L’endoscope
traditionnel est le dernier appareil
médical invasif qui ne peut pas être
stérilisé.
·
Les protocoles de
retraitement (intervention
humaine) ne garantissent pas une désinfection à 100 %.
·
Des cas de
contamination graves après des procédures
endoscopiques surviennent chaque année.
·
Des rappels de
patients pour des examens bactériens
approfondis après des procédures endoscopiques (250.000
patients par an aux
USA, ce qui représente environ 1 % de la totalité des
patients).
·
En France, le don du
sang est impossible après un
examen endoscopique.
·
Exposition à
des maladies et infections inconnues à
découvrir au cours des mois ou années à venir.
Pour le praticien et les centres
hospitaliers :
·
Augmentation des
plaintes juridiques contre les
praticiens et les centres hospitaliers.
·
Augmentation des
coûts d’assurances pour les
praticiens et les centres hospitaliers.
·
Un personnel
important chargé du retraitement des
endoscopes.
·
De nouveaux
protocoles de retraitement tous les 6
mois, nouvelles formations pour le personnel.
·
Les nouveaux
protocoles de retraitement augmentent
le coût des endoscopies.
·
Tous les protocoles
de retraitement exigent une
documentation / traçabilité qui nécessite beaucoup
de temps.
·
L’utilisation de
nouveaux produits chimiques (APA)
détériorent rapidement la matière des appareils
(augmentation de la maintenance
de 30 %)
·
Investissement dans
des appareils de retraitement
coûteux et complexes (laveuses).
·
L’endoscopie
d’urgence est impossible ou limitée à
la disponibilité du personnel.
·
Impossibilité
de réaliser des endoscopies à
l’extérieur des centres hospitaliers.
·
La réalisation
d’endoscopies au domicile du client est
difficile, voir impossible.
·
La réalisation
d’une endoscopie sur un patient avec
CJD (Maladie de Creutzfeldt-Jakob) est impossible.
·
La destruction des
endoscopes utilisés sur des
patients avec CJD est nécessaire.
·
Le risque de rappels
de patients et d’examens
supplémentaires coûteux pour s’assurer qu’ils n’aient pas
été contaminés.
Pour la société :
·
Les protocoles de
retraitement d’endoscopes
complexes augmentent significativement les dépenses
médicales.
·
Les produits
chimiques de retraitement utilisés actuellement
sont-ils efficaces contre des infections encore inconnues ?[4]
3.3 Les
solutions d’Axess Vision Technology
Figure
6 Processus
choisi par Axess Vision Technology
Les fondateurs ont
étudié différentes voies techniques qui pourraient
simplifier l’utilisation des endoscopes flexibles afin de supprimer les
risques
élevés de contaminations croisées. Les discussions
avec des fournisseurs de
lentilles d’objectifs ont déclenché le lancement
technique du projet, offrant
des opportunités potentielles pour un endoscope jetable. Les
recherches ont été
orientées vers une nouvelle génération
d’endoscopes pour limiter ou supprimer
les désavantages actuels des endoscopes médicaux
existants.
Les spécifications
définies pour cette nouvelle génération
d’endoscopes
se présentent ainsi :
·
Un dispositif
d’imagerie de qualité
·
Un appareil
d'interface utilisateur convivial
·
Un dispositif simple
·
Des
caractéristiques ergonomiques équivalentes à
celles
des équipements traditionnels
·
Un dispositif
entièrement jetable
·
Un dispositif peu
coûteux
Les capacités et les
combinaisons de ces différentes nouvelles
technologies ont satisfait aux exigences du marché en permettant
la conception
d’un endoscope jetable simple et peu coûteux, afin de supprimer
les problèmes
de contaminations croisées et de restrictions de budgets.
Le tube d’insertion DispoFlex
se caractérise par :
·
Un tube d’insertion
stérile et jetable en matières
plastiques biocompatibles,
·
Un circuit de canaux
d’air / d’eau stérile et
jetable,
·
Une poignée de contrôle réutilisable sur
laquelle s’ajoutent un tube d’insertion et le circuit air/eau
garantissant une
stérilité absolue,
·
Un capteur
d’images numérique breveté et son logiciel d’analyse pour
l’amélioration
automatique de la luminosité,
·
L’utilisation
de lentilles en plastique spécialement conçues à
cet effet,
·
Une base
propriétaire comprenant une carte d’acquisition via un PC, une
pompe de succion
et des systèmes de commande à pédales,
·
Un logiciel
d’analyse et de gestion de données de patients qui gère
les textes, photos et films.
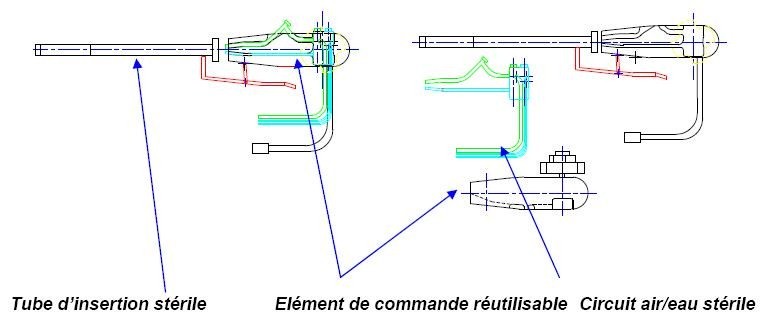
Figure 7
DispoFlex, Schéma global

Figure 8 Ensemble des éléments de l'endoscope
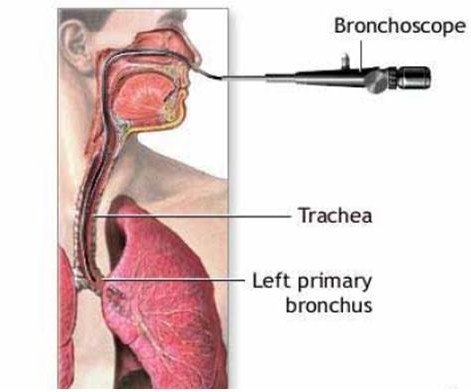
Figure
9 Voie de
passage d'un bronchoscope
retour sommaire
DispoFlex est un
dispositif économique qui améliore la productivité
des utilisateurs. Il évite
les étapes de retraitement coûteuses et ne
nécessite plus de personnel
spécialisé,
plus de traçabilité complexe, plus de produits chimiques,
plus de laveuses
automatiques, plus de locaux de désinfection
spécialisés, plus de salles
d’endoscopie spécialisées, plus de frais de maintenance,
plus de temps d’arrêt.[4]
L’objectif de ce stage
est de répondre
aux exigences essentielles de la norme ISO 13485 pour qu’Axess Vision
Technology accède à la certification d’ici fin 2009.
Axess Vision Technology travaille avec le cabinet
de
consultants CEISO. Une réunion mensuelle à laquelle CEISO
apporte son soutien
aussi bien au département des affaires réglementaires
qu’au département qualité
est faite au sein d’AVT.
Pour avoir une bonne vision de la mission, et d’en
déterminer les objectifs, plusieurs outils découverts
lors des ateliers de la
formation du master de management de la qualité ont
été utilisés, tels que le
QQOQCP, la note de clarification, la planification dynamique
stratégique (PDS).
Ceux-ci ont permis de déterminer une façon de
procéder pour la résolution de la
problématique. [6]
1-
Etat des lieux de la mission
Afin de prendre
connaissance de
la mission de stage et d’identifier sa problématique, une note
de clarification
a été réalisée, comme ci après :
Mettre
en place et intégrer un système qualité qui
répond aux
référentiels ISO 9001 et ISO 13485
CONTEXTE
Axess Vision Technology, Start-up
médicale labellisée
« entreprise innovante », développe de
nouveaux instruments dans le
domaine de l'endoscopie médicale.
L’objectif principal est de garantir une mise en
place
rapide du système de management de la qualité, de
l’Annexe II de la directive
93/42/CEE, des exigences du CMDCAS et de celles de la FDA (CFR 21 PART
820).
Il s’agira entre
autres de
constituer l’essentiel de la base documentaire et d’apporter une
efficacité
maximale à l’organisation en place.
DONNEE
D’ENTREE
Convention de stage
Normes ISO
9001 et ISO
13485
OBJET DU
PROJET
Rédiger les procédures et documents
du système qualité
Mettre en place le système qualité
Intégrer le
système qualité
PRODUIT
DU PROJET
Système qualité opérationnel
Système
qualité optimum pouvant
obtenir la certification ISO 13485.
OBJECTIFS
VISES
Obtenir la
certification ISO
13485 avant décembre 2009.
ACTEURS
DU PROJET
Demandeurs : Olivier FRUCTUS, Nicolas MATHIEU
Maître d’œuvre : Anne Hermelin
Suiveur UTC :
Gilbert
Farges
CONSEQUENCES
ATTENDUES
Obtention de la certification en vue de
l’obtention du
marquage CE en 2009.
Mise sur le
marché hospitalier
des endoscopes jetables en 2010.
CONTRAINTES
DU PROJET
Aucunes
données d’entrées sur
lesquelles fonder mon travail
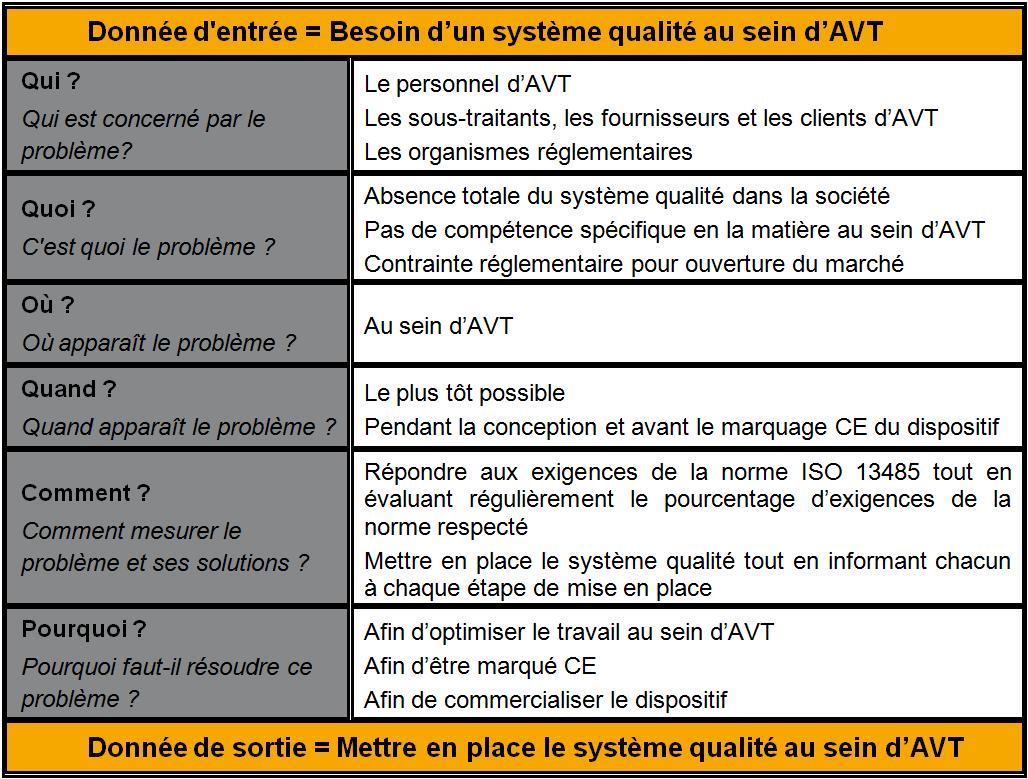
2- Clarification
de la problématique
Afin d’être
certain que la
problématique est bien cernée, il est important de
prendre en compte tous les
aspects de la mission. Le QQOQCP présenté ci-après
est l’outil utilisé pour
préciser la mission à réaliser.
Figure 10 QQOQCP
retour sommaire
La Planification Dynamique Stratégique
permet de déterminer
clairement la nécessité de mettre en place le
système qualité répondant à la
norme ISO 13485.
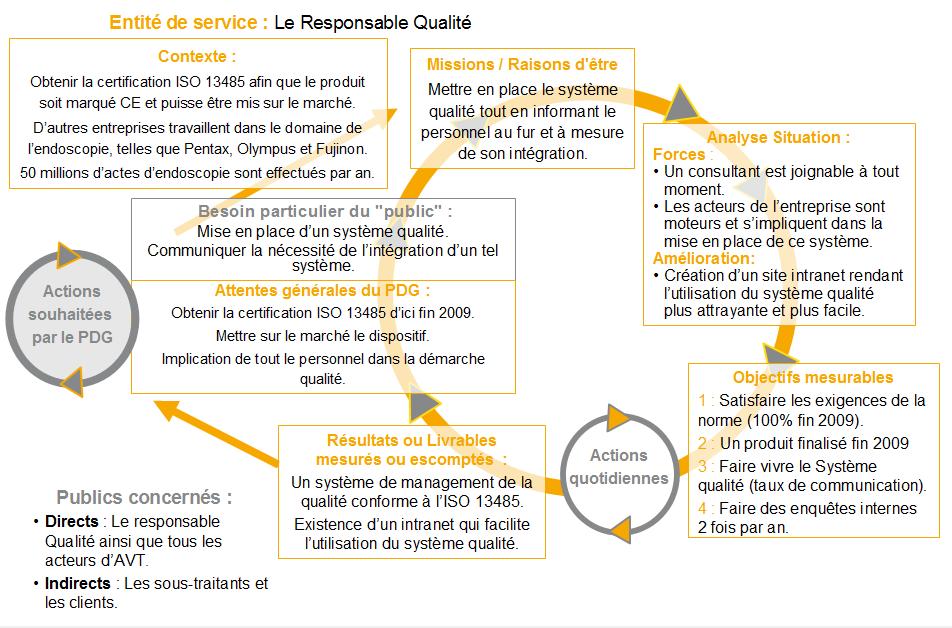
Figure
11
Planification Dynamique Stratégique
4- Méthodologie
de travail
Pour mener la mission à bien, la
méthode du processus accéléré
de résolution de problèmes a été choisie.
Elle est décrite ci-après.
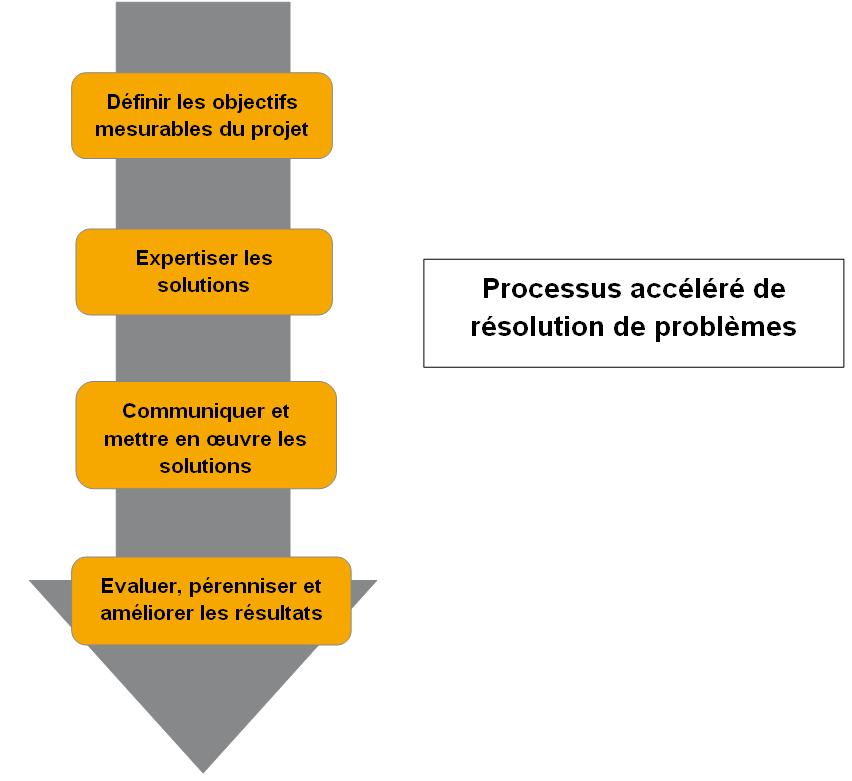
Définir
les objectifs
mesurables du projet :
·
Définir
les missions du projet et les mesures de succès.
·
Documenter
le projet. Collecter les données et les informations qui y sont
relatifs.
·
Analyse
du besoin de l’entreprise : collecter les informations et les
données.
·
Contrôler les missions :
s’assurer que le
client et moi même sommes d’accord sur la problématique et
les besoins.
Documents produits :
·
Processus de la
société.
·
Etat des lieux.
·
Note de clarification.
·
Méthodologie de travail.
·
Planning prévisionnel
retour sommaire
Expertiser
les
solutions :
·
Développer
les solutions.
·
Analyser
les solutions et les options d’intervention développées.
·
Choisir une option d’intervention et
élaborer le
plan d’intervention.
Documents produits :
Comptes rendus des réunions avec le
consultant CEISO.
Communiquer
et mettre en
œuvre les solutions
·
Communiquer
les solutions.
·
Mettre
en place le plan d’intervention.
·
Piloter la mise en place de
l’intervention.
Documents produits :
·
Livrables.
·
Mails
d’informations.
·
Affichages.
·
Mise
en place du système qualité.
Evaluer,
pérenniser et
améliorer les résultats:
·
Solliciter
l’avis de chacun.
·
Revoir et améliorer les
documents mis en place.
Documents produits :
·
Livrables.
·
Echanges de mails.
5-
Planification de
différentes
actions à accomplir
Afin
de couvrir
l’ensemble des éléments à mettre en place et de ne
pas en oublier, le rétro
planning suivant la méthode de travail choisie a
été établi.
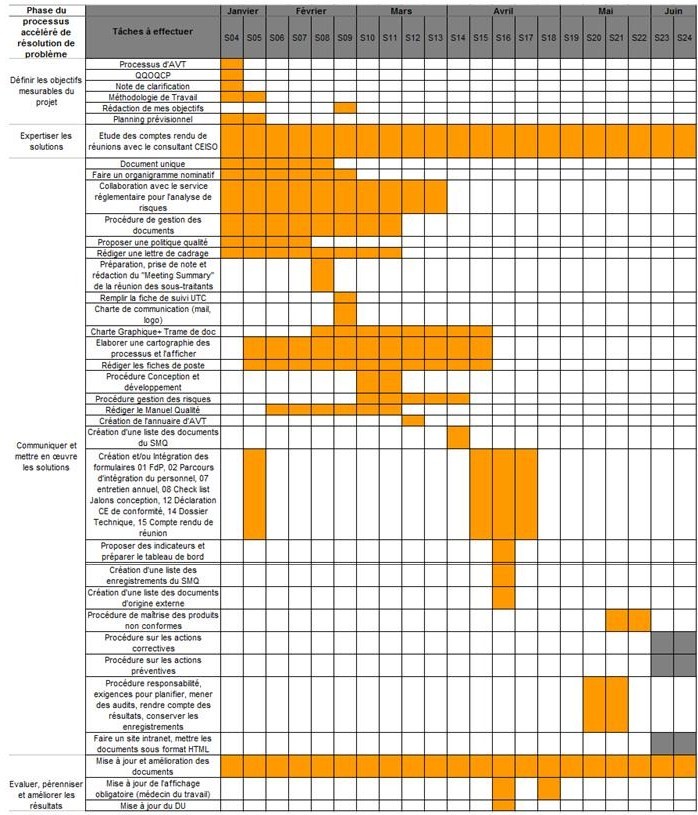
Figure
12 Rétro planning du déroulement du stage
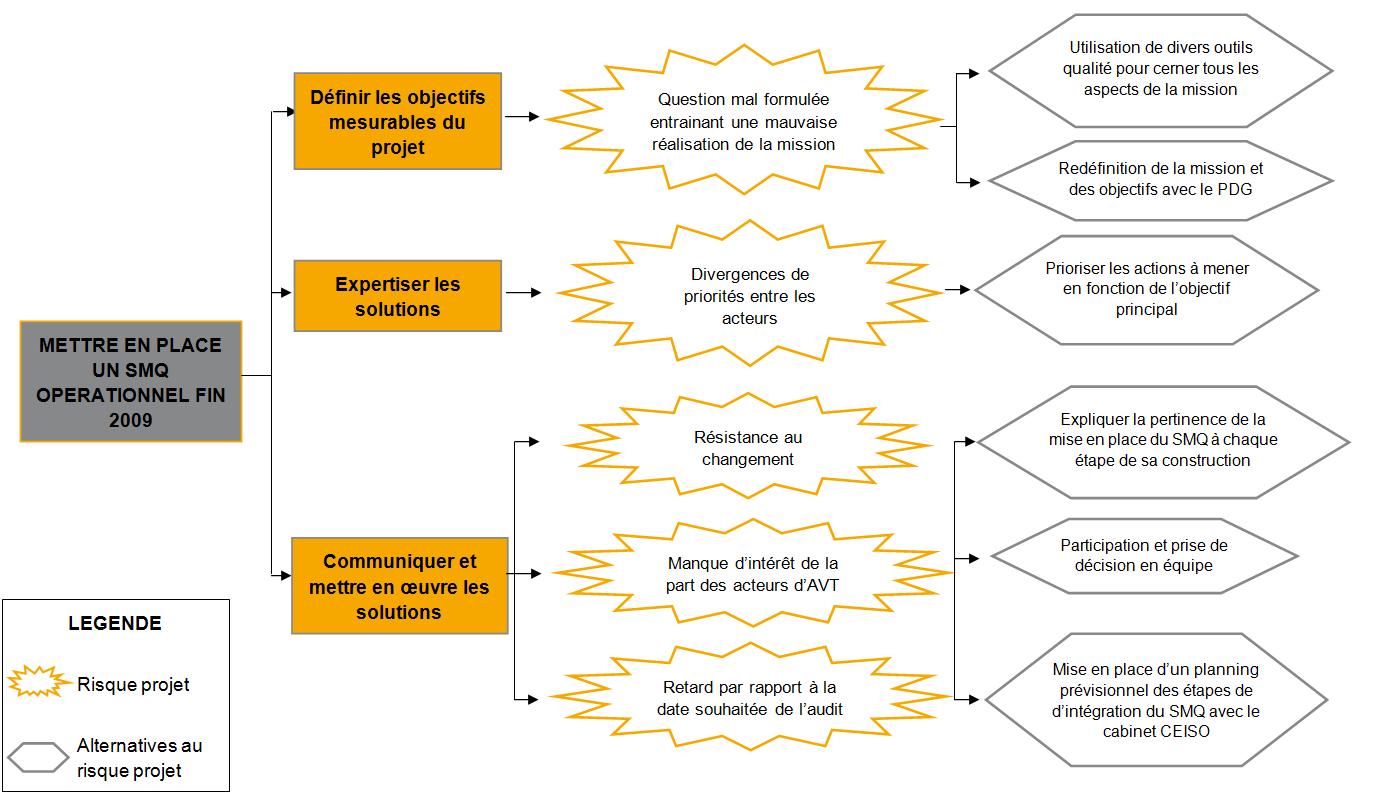
Figure
13
Diagramme de décisions
1-
Résultats
1.1 La Cartographie de
processus
Axess Vision Technology est une start-up, le
processus
d’intégration de la qualité a débuté avec
le début de ce stage.
La première chose indispensable à
mettre en place est la
cartographie des processus.
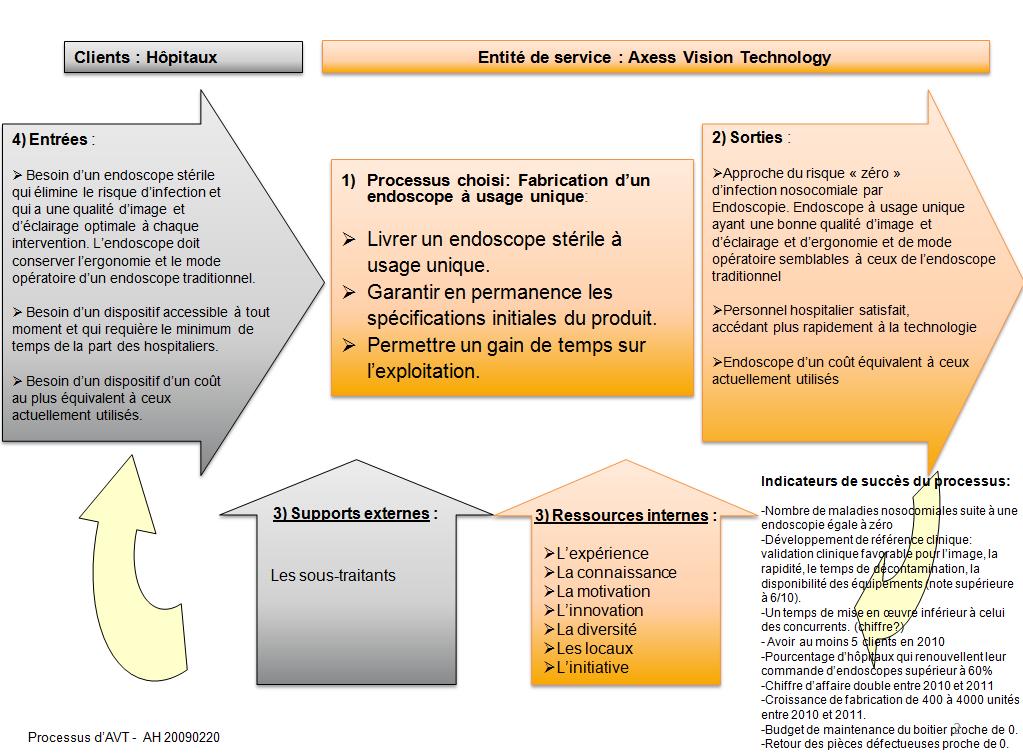
Pour la réaliser, 4
processus ont été identifiés
PROCESSUS
: CONCEVOIR ET
DEVELOPPER DES PRODUITS ET DES SERVICES
Finalité : Concevoir et
développer des dispositifs et
des services en conformité avec les exigences
réglementaires
·
avec
un prix de revient permettant de générer un Chiffre
d’Affaire et une
rentabilité suffisante
·
dans le respect des budgets et des
plannings de
conception prévus.
PROCESSUS
: PRODUIRE ET
CONTRÔLER
Finalité : Produire :
·
les
quantités de dispositifs ou accessoires satisfaisant les besoins
des clients et
de l'entreprise
·
en
conformité avec la documentation technique marquage CE
·
livrés
dans les temps
·
à des conditions de
coûts acceptables
PROCESSUS
: PROMOUVOIR ET
COMMERCIALISER LES DISPOSITIFS MEDICAUX D’AVT
Finalité :
·
Satisfaire
les besoins des clients en proposant les produits et services de
l'entreprise
·
Satisfaire les objectifs de vente de
l'entreprise pour assurer sa rentabilité et sa
pérennité et fournir les marges
de manœuvre pour investir et préparer l'avenir et distribuer une
partie des
profits aux actionnaires et aux collaborateurs
PROCESSUS
: CORRIGER ET
AMELIORER DE MANIERE CONTINUE
Finalité : Surveiller les
performances des processus
et du SMQ, veiller à la conformité des produits et des
services, traiter les
retours des clients
Analyser les
données et Proposer
et/ou Suivre la mise en œuvre et l'efficacité des actions
correctives,
préventives et d'amélioration.
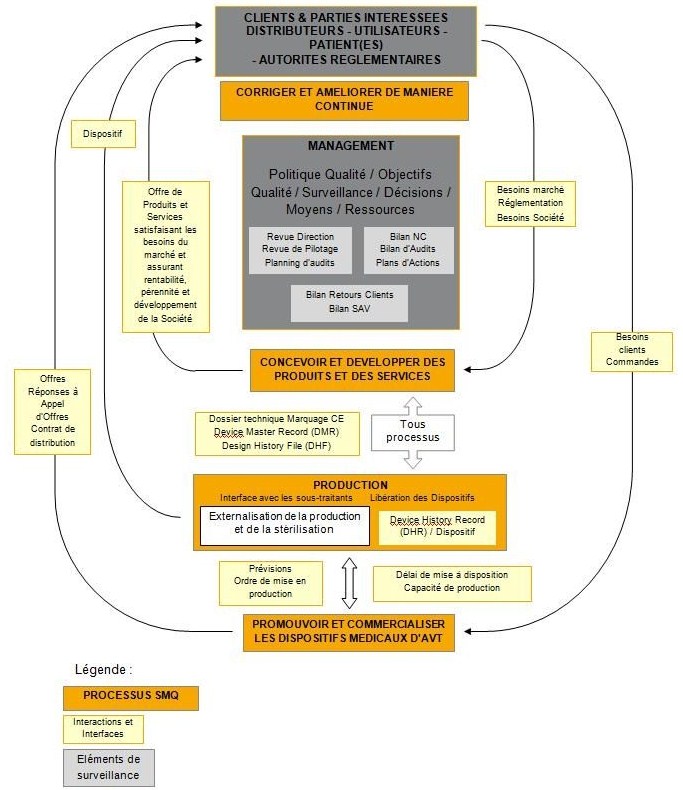
Figure 14 Cartographie des processus
1.2
La politique Qualité et la lettre de cadrage
Afin de montrer l’engagement de la direction dans
la qualité
et le début de l’intégration de la qualité dans
l’entreprise, une politique
Qualité a été rédigée et les
objectifs ont été déclinés dans la lettre
de
cadrage.

Figure
15
Politique Qualité

Figure
16 Lettre de
Cadrage
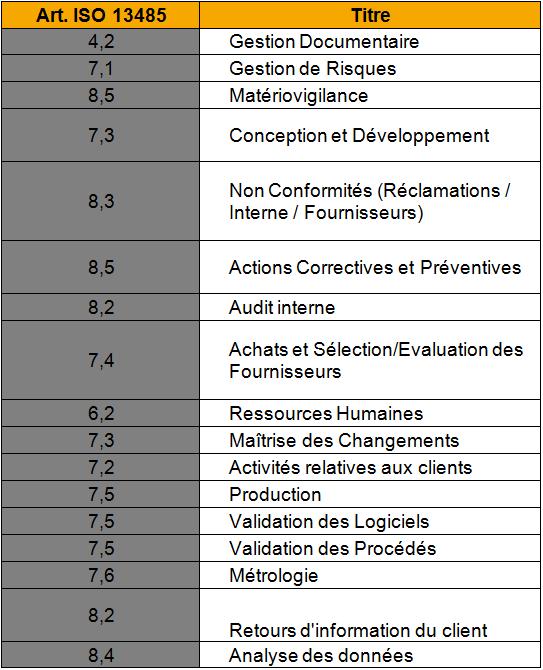
1.3 Les procédures
Les 17 procédures listées ci-dessous
sont mises en place pour
répondre aux exigences de la norme ISO 13485. A ces
procédures sont rattachés
des formulaires. Ceux-ci sont intégrés dans le
système en même temps que la procédure
qui y fait référence. (Cf Annexe 2 procédure 1
Gestion documentaire).
Tableau 1 Procédures répondant
aux exigences de la norme ISO 13485
1.4 Le Tableau de Bord
Le tableau de bord permet de visualiser les
indicateurs
choisis pour chaque processus. Lors d’une des réunions mensuelle
avec le
cabinet CEISO, nous avons pris le temps de choisir les indicateurs les
plus
pertinents pour chaque processus, puis les objectifs à atteindre
en 2009 ont
été fixés.
Actuellement en phase de conception du dispositif,
le
tableau de bord est mené à évoluer très
rapidement, dés que la phase de vente
est entamée.
Ce tableau de bord est revu et
complété tous les 3 mois. Il
est important, pour assurer sa survie et son utilité, de le
revoir assez
fréquemment dans le début de sa mise en place. Cela
permet de le faire évoluer
en cas de besoin et de faire en sorte qu’il s’adapte au plus
près des besoins
actuels de la société
Il sera
édité pour chaque revue
de direction, afin de faire un point sur le statut des objectifs. Un
compte
rendu de réunion statuera sur les actions à mener.
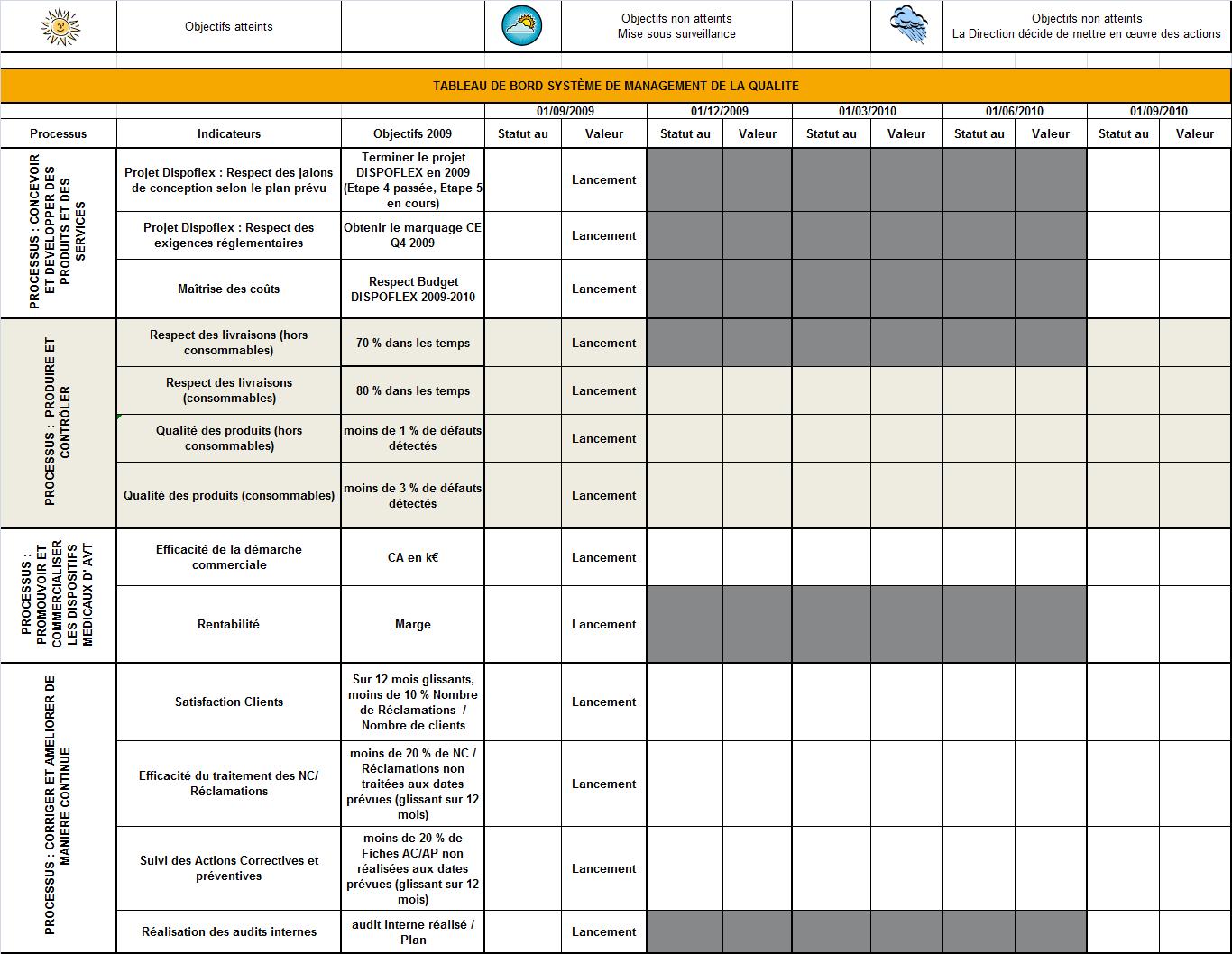
Tableau 2 Tableau de bord
retour sommaire
1.5 Le Manuel Qualité
Le Manuel Qualité peut être
élaboré sous diverses formes.
Axess Vision Technology étant une société
débutante dans la qualité, il semble
judicieux de le rédiger suivant le plan de la norme ISO 13485.
(Cf Annexe 3)
De cette manière, nous sommes sûrs de
répondre à l’ensemble
des exigences imposées par la norme, et il est ainsi plus
aisé, lors de
l’audit, de prouver que tous les points de la norme ont
été pris en compte.
2-
Discussion et perspectives
Un autodiagnostic sur les exigences de l'ISO 13485
est
actuellement en cours de réalisation. Il est basé sur le
fichier Excel déjà
disponible sur l’internet du master MQ qui permet de réaliser
l’autodiagnostic
sur les exigences de la norme ISO 9001. La norme ISO 13485 repose sur
la norme
ISO 9001 et comporte en plus des exigences relatives aux dispositifs
médicaux.
Le travail indispensable avant son utilisation est de le mettre
à jour et de le
compléter avec les exigences essentielles spécifiques
à la norme ISO 13485.
Réalisé dans le mois qui suit, il sera d’une grande
utilité pour statuer sur le
pourcentage d’exigences de la norme satisfaites et planifier une date
pour
l’audit.
AVT étant une jeune entreprise, son manuel
qualité suit
scrupuleusement le plan de la norme ISO 13485. Une fois la
certification
obtenue et une expérience prolongée de la qualité
par les acteurs de la
société, le Manuel Qualité pourra être
repensé et rédigé sous une autre forme.
Une fois les documents relatifs à la
qualité validés, un
site intranet sera élaboré. L’objectif de ce projet est
de rendre le système
qualité plus attractif, intuitif est donc simple d’utilisation.
En effet, à mon
arrivée, la qualité n’était pas connue de
l’ensemble des acteurs. Par ce biais,
nous espérons que ces acteurs parcourront sans
« crainte » le système
qualité.
Bilan
de
la mission
En
collaboration avec le PDG d’AVT, les objectifs de mon stage ont
été définis au
début de ma mission, en janvier 2009. Aujourd’hui, en juin 2009,
il est
possible de faire un bilan sur le pourcentage de réalisation des
objectifs.
Tableau
3 Bilan
des missions réalisées
Apport
à l’entreprise
Ma mission au
sein d’AVT porte quotidiennement ses fruits. Le point le plus marquant
au terme
de ces 5 mois est la volonté que chacun a de vouloir s’impliquer
dans la
qualité et de vouloir faire fonctionner le système. La
mise en place de la base
du système qualité est en phase d’achèvement,
l’objectif suivant est de faire
vivre le système qualité. L’audit est prévu pour
fin décembre 2009. Il nous
implique tous et nous œuvrons ensemble pour atteindre cet objectif.
Apport
personnel
Sur les appuis
des notions vues en cours au semestre d’automne 2008/2009 à
l’UTC, ce stage a eu
pour objectif de construire un système qualité. Celui-ci
affermit et complète
par son côté pratique la formation théorique de
management de la qualité reçue.
En effet, confrontée
à la réalité de la vie en entreprise, j’ai
réalisé qu’il n’était pas toujours
évident de programmer des réunions à un moment
opportun pour chacun. J’ai donc appris
à tenir compte des disponibilités de chacun pour fixer
des dates de réunions
d’avancement de projet ou de validation, et à savoir
redéfinir les priorités.
Le manque de
disponibilité de mon maître de stage et la confiance qu’il
m’a accordée m’ont
fait évoluer dans ma capacité à travailler en
forte autonomie. Ceci me conforte
dans mon désir de travailler dans le domaine managérial
et en collaboration
avec les différents acteurs de l’entreprise.
retour sommaire
AAGR : Average
Annual Growth Rate
AFNOR:
Association Française de
Normalisation
AVT : Axess Vision Technology
CCD :
Charge-Coupled
Device, ou dispositif à transfert de charge
CJD : Maladie de
Creutzfeldt-Jakob
CMDCAS :
Canadian Medical Devices
Conformity Assessment System
DM : Dispositif
Médical
FDA : Food
and Drug Administration
ISO :
International Organization for Standardization
MQ : Manuel
Qualité
PDG : Président-Directeur
Général
PDS : Planification
Dynamique Stratégique
QQOQCP : Qui
Quoi Où Quand Comment Pourquoi
SMQ : Système de Management
de la Qualité
UTC : Université de
Technologie de Compiègne
510K : Au moins 90 jours avant qu’il soit
commercialisé pour la
première fois le dispositif médical doit être
soumis à une « Notification de
pré-marché », aussi appelée «
Soumission » 510(k). Celle-ci doit contenir des
informations suffisantes démontrant que le dispositif est
substantiellement
équivalent à un dispositif préalable;
c’est-à-dire, un dispositif déjà
légalement commercialisé.
Une
Notification de pré-marché est aussi requise pour les
produits, déjà légalement
mis sur le marché ou qui le seront ultérieurement,
lorsqu’ils ont été ou vont
être considérablement modifiés, de façon
à assurer leur sécurité et efficacité. Présentation
d’Axess Vision
Technology [7]
CEISO : Organisme de conseil, audit et formation dans le
domaine réglementé
des dispositifs médicaux, CEISO dispose en interne de plusieurs
compétences et
expériences complémentaires de manière à
répondre au mieux à l’ensemble des
problématiques auxquelles les entreprises peuvent être
confrontées. [8]
Marquage
CE : Inscription
apposée
de façon visible sur un équipement mis sur le
marché soit directement, soit sur
une fiche signalétique. Il est apposé par le fabricant qui garantit ainsi que l’équipement est conforme
aux exigences
essentielles
de la Directive équipement sous pression (DESP).
Start-up :
Société en construction qui ne s’est pas encore
lancée sur le marché
commerciale
Les
référentiels normatifs
[1] ISO 9000
: Système de management de la qualité – principes
essentiels et vocabulaire, Ed AFNOR, 2005, 38p, ICS 01.040.03.
[2] ISO
13485 : Systèmes de management de la qualité –
Exigences à des fins réglementaires, Ed AFNOR, 2004, 75p,
ICS : 03.120.10.
[3] ISO
62 366 : Medical
devices – Application of usability engineering to medical devices, Ed
AFNOR, 2007,
215p.
Ouvrage interne
[4]
AVT_BP_OF20080501: Business Plan : Innovation dans l’Endoscopie
Souple, Olivier Fructus, 2008, 58p.
Les sites
consultés
[5] Site de
Futura-Sciences – Magazine de l’innovation, de la
science et de la santé, www.futura-sciences.com,
consulté le 12 mai 2009.
[6] Site du
master MQ de l’UTC – mis à jour par G. Farges ; www.utc.fr/mastermq
consulté de janvier à
juin 2009.
[7] Site de
Matrix Medical Consulting Corporation - société
professionnelle de consultation et de recherches contractuelles
spécialisées
dans le domaine de la réglementation, des essais cliniques et de
l'assurance de
la qualité pour les industries biotechnologiques,
pharmaceutiques et des
appareils biologiques à l'échelle mondial ;
www.matrixmedcorp.com
consulté le 19 mai 2009.
[8] Site de CEISO – www.ceiso.fr
consulté le 16 avril 2009.
ANNEXE
1 : Liste des lois pour la définition des
spécifications réglementaires.
|
Pays
|
Réglementation
|
N
|
Spécifications
|
|
France
|
Code de la Santé
Publique transposant la Directive 93/42/CEE
|
REG 1-1
|
Un Dossier Technique (Cf. Annexe VII de la
Directive 93/42/CEE) décrit le Dispositif, son usage, etc. et
démontre la conformité aux exigences essentielles de
l'Annexe I de la Directive 93/42/CEE.
|
|
REG 1-2
|
AVT (Fabricant) dispose d'une ou des
attestation(s) CE d'un Organisme Notifié.
|
|
REG 1-3
|
Si ce n'est pas déjà le cas,
un correspondant matériovigilance a été
désigné pour AVT à l'AFSSAPS
|
|
REG 1-4
|
Si le dispositif médical (DM) est de
Classe I, une déclaration avant la mise sur le marché
doit être faite auprès de l'AFSSAPS.
|
|
REG 1-5
|
Si le DM Classe IIb et III, une
Déclaration avant la mise sur le marché doit être
faite auprès de l'AFSSAPS conformément au décret
2002-1221.
|
|
France
|
Code de l'Environnement
(Décret 2005-829)
|
REG 2-1
|
L'étiquetage du Dispositif comporte
le logo "Poubelle barrée"
|
|
REG 2-2
|
La notice d'utilisation mentionne,
conformément au décret 2005-829 transposant la directive
DEEE, les modalités pour l'élimination et le recyclage du
dispositif.
|
|
USA
|
·
Federal Food,
Drug and Cosmetic Act
·
CFR 21 Part 800 to 1299
·
CFR 21 Part 1 to 99
|
REG 3-1
|
AVT est enregistré auprès de
la Food, Drug Administration FDA.
|
|
REG 3-2
|
AVT a listé ses produits
auprès de la FDA.
|
|
REG 3-3
|
Sauf si le Dispositif est exempté,
AVT a obtenu de la FDA la SE Letter (510(k)) ou le PMA.
|
|
REG 3-4
|
Les Dispositifs de AVT sont conçus,
développés, fabriqués et maintenus selon le CFR 21
Part 820.
|
|
Canada
|
Règlements des
Instruments Médicaux (RIM)
|
REG 4-1
|
AVT dispose d'une homologation (Classe II,
III ou IV)
|
|
REG 4-2
|
AVT dispose d'un certificat ISO 13485
émis par un Registraire (Classe II, III ou IV)
|
|
REG 4-3
|
AVT dispose d'un contrat de distribution
avec un distributeur sur le territoire Nord Américain qui
possède une licence d'établissement adéquate pour
distribuer les dispositifs d’AVT
|
|
Allemagne
|
Idem qu’en France +
Spécificités éventuelles
|
REG 5-1
|
Les notices d'utilisation et
l'étiquetage sont traduits dans la langue du pays. Cette
traduction a été validée par une personne
possédant la double compétence "Utilisateur" et
linguistique.
|
|
REG 5-2
|
AVT dispose d'un contrat de distribution
avec un distributeur qui possède les autorisations locales
adéquates pour distribuer les dispositifs d'AVT
|
|
REG 5-3
|
Si nécessaire, le distributeur a fait
la déclaration de mise sur le marché auprès des
autorités compétentes du pays.
|
ANNEXE 2 : PR01 :
GESTION
DOCUMENTAIRE
|
|
Rédacteur
|
Vérificateur
|
Approbateur
|
|
Nom :
|
AH
|
OF
|
OF
|
|
Fonction :
|
Responsable Qualité
|
PDG
|
PDG
|
|
Date :
|
09/03/2009
|
|
|
|
Signature :
|
|
|
|
|
SUIVI DES MODIFICATIONS
|
|
Version
|
Date
|
Rédacteurs
|
Observations
|
|
01
|
09/03/2009
|
AH
|
Création du document
|
|
|
|
|
|
OBJET
Cette
procédure décrit les
dispositions mises en œuvre pour la maîtrise des documents, des
données et des
enregistrements.
DEFINITIONS
Procédure
(P) :
Document décrivant la manière d’accomplir une
activité, un service ou un
processus.
Mode
opératoire (MO) : Manière
spécifiée
d’effectuer un enchaînement d’opérations ou de
tâches.
Enregistrement
(E) : Document faisant
état de résultats obtenus ou apportant la preuve de la
réalisation d’une
activité.
Consigne
(C) : Support d’information
spécifiant l’exécution d’une opération ou d’une
tâche dans un cadre bien précis.
Formulaire
(F) : Document
pré-formaté servant à enregistrer les informations
ou actions
réalisées au cours d’une activité.
Vérification :
La vérification vise à
s‘assurer de la conformité du contenu du document
vis-à-vis des exigences
Qualité.
Approbation :
L’approbation consiste à valider les informations contenues
dans un
document et permet sa diffusion.
Diffusion :
Modalités de mise à
disposition des documents auprès des utilisateurs.
Classement :
Opération qui permet de
ranger les documents ou enregistrements pendant le temps où ils
sont utilisés
ou renseignés.
Archivage :
Opération qui permet de conserver les documents ou
enregistrements une fois
utilisés ou périmés afin
d’en assurer
l’historique.
|
820.181 Dossier Maître
du dispositif (Device Master Record DMR)
Tout fabricant doit tenir à jour
des DMR (Dossier de Maître du Dispositif). Tout fabricant doit
assurer que tout DMR est préparé et approuvé en
accord avec le §820.40. Le DMR pour chaque type de dispositif doit
inclure ou se référer à l’emplacement de
l’information suivante :
a)
Les
spécifications de dispositif comprenant les schémas
appropriés, la composition, la formulation, les
spécifications de composants et les spécifications de
logiciel.
b)
Les
spécifications de processus de production, y compris les
spécifications d’équipements appropriés, les
méthodes de production, les procédures de production et
les spécifications d’environnement de production.
c)
Les
procédures et spécifications d’assurance de la
qualité y compris les critères d’acceptation et
l’équipement d’assurance de la qualité devant être
utilisés, et
d)
Les
spécifications d’emballage et d’étiquetage, y compris les
méthodes et les processus utilisés.
e)
Les
méthodes et procédures pour l’installation, la
maintenance et les activités de services.
|
|
820.184 Dossier Historique de
Production d’un dispositif (DHR)
Tout fabricant doit tenir à jour
des DHR (Dossier Historique de production d’un Dispositif). Tout
fabricant doit établir et tenir à jour des
procédures pour assurer que les DHR pour chaque groupe, lot ou
unité sont tenus à jour pour démontrer que le
dispositif est fabriqué selon le DMR et les exigences de cette
section.
Le DHR doit comprendre ou faire
référence à l’emplacement des informations
suivantes :
a)
La date de
fabrication.
b)
La
quantité fabriquée.
c)
La
quantité mise en circulation pour distribution.
d)
Les
enregistrements d’acceptation qui démontrent
que le dispositif est fabriqué selon le DMR.
e)
L’étiquette
et l’étiquetage d’identification primaires utilisés pour
chaque unité de production, et tout numéro(s)
d’identification et de contrôle utilisé.
|
|
820.186 Enregistrement du
Système Qualité (QSR)
Tout fabricant doit tenir à jour
les enregistrements du Système Qualité (QSR).
Le QSR doit comprendre ou se
référer à l’emplacement des procédures et
la documentation des activités exigées par cette section
qui ne sont pas spécifiques à un type particulier de
dispositif(s), y compris, mais sans y être limité, les
enregistrements exigés par le §820.20.
Tout fabricant doit assurer que le QSR est
préparé et approuvé selon les termes du
§820.40.
|
|
820.30 j) DHF Dossier
Historique de Conception.
Tout fabricant doit établir et
tenir à jour un DHF pour chaque type de dispositif. Le DHF doit
contenir ou référencer les enregistrements
nécessaires pour démontrer que la conception a
été développée selon le plan de conception
approuvé et les exigences de cette section.
|
REFERENTIELS
§
4.2.3 & 4.2.4 NF EN ISO 13485:2004, § 4.2.3 & 4.2.4 NF EN
ISO
9001 : 2000
Directive
93/42/CEE
http://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=CELEX:31993L0042:EN:HTML
CFR 21
Part 820.40 Maîtrise des documents (Document Control)
CFR 21
Part 820.180 Enregistrements- Exigences générales (Record
General
Requirements)
CFR 21
Part 820.181 Dossier Maître du Dispositif (Device Master Record -
DMR)
CFR 21
Part 820.184 Dossier Historique de Production (Device History
Record - DHR)
CFR 21
Part 820.186 Enregistrement du système Qualité (Quality
System
Record - QSR)
CFR 21
Part 820.30 j. Dossier Historique de la Conception (Design
History File – DHF)
http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?CFRPart=820
Medical Device Quality
Systems Manual FDA 97-4179 (December 1996) http://www.fda.gov/cdrh/dsma/gmpman.html
Maîtrise
des documents et des Enregistrements
Les
dispositions pour la maîtrise des documents et des
enregistrements
comprennent :
·
La
revue et l’approbation des documents quant à leur
adéquation avant diffusion.
Cela se manifeste par les noms des personnes désignées
pour chaque fonction
dans les tableaux en début de document, de la date et de la
signature. Pour les
documents avec les signatures du rédacteur, du
vérificateur et de
l’approbateur, la signification des signatures est la suivante :
º
Le
rédacteur atteste de la complétude du document et qu’il
est prêt pour la revue.
Il est généralement choisi pour son expertise, sa
connaissance et son
implication dans les activités concernées par le
document.
º
Le
vérificateur atteste de la pertinence du fond et de la forme. Il
est
généralement choisi pour les mêmes raisons que le
rédacteur et selon
l’importance du document pour son positionnement hiérarchique.
º
L’approbateur
vérifie que ce sont des personnes désignées et
compétentes qui ont réalisé et
vérifié le document. Le positionnement
hiérarchique dépend de l’importance du
document (Direction Générale pour un document
général (Manuel Qualité ou
Procédures générales par exemple) ou Responsable
de la fonction (ou du service)
concernée pour un document plus ciblé).
La
revue et l’approbation ne sont pas
nécessairement disjointes et peuvent être confondues.
·
La
revue, mise à jour si nécessaire et
nouvelle approbation des documents.
·
L’identification
des modifications sur les
documents (historique en début de document) et de la version
selon le modèle
suivant :
|
SUIVI DES MODIFICATIONS
|
|
Version
|
Date
|
Rédacteurs
|
Observations
|
|
01
|
09/03/2009
|
AH
|
Création
du document
|
|
|
|
|
|
|
|
|
|
|
·
Des Listes
récapitulatives
afin de connaître les documents et la version en vigueur (L01
pour les documents
du SMQ et L DOC EXT pour les documents d’origine externe).
·
La disponibilité
sur les
lieux de l'utilisation des versions pertinentes des documents
applicables (une
version papier de référence, et diffusion par le
réseau informatique en Lecture
Seule).
·
L’assurance que les
documents restent lisibles et facilement identifiables.
·
L’assurance que les
documents d'origine extérieure sont identifiés et que
leur diffusion est
maîtrisée (par exemple, les normes,
réglementations,…).
·
L’identification de
manière
adéquate des documents périmés pour éviter
toute utilisation non intentionnelle
(document barré, date).
·
L’assurance que les modifications apportées aux documents
sont revues et approuvées soit par l'autorité
d'approbation d'origine, soit par
toute autre autorité désignée ayant accès
à des informations connexes
pertinentes lui permettant d'étayer sa décision (Ce sont
en fait les mêmes
fonctions qu’à l’origine qui interviennent dans la modification).
·
La définition de la
période
pendant laquelle une copie au moins
des documents maîtrisés périmés est
conservée (Voir L02 Liste des
enregistrements). Cette période permet d’assurer que les
documents conformément
auxquels les dispositifs médicaux ont été
fabriqués et soumis à l'essai sont
disponibles pendant une durée au moins égale à la
durée de vie du dispositif
médical définie (Voir ci-dessous).
Des
modifications
temporaires de manière manuscrite (avant révision
formelle) ou permanentes sur
les enregistrements sont possibles sur les versions papier. Elles
doivent être
accompagnées de la signature de la personne compétente
qui effectue cette
modification et de la date.
Par
exception, les Listes récapitulatives comme L01, L02,… sont des
documents très
évolutifs et la revue et approbation est réalisée
par le RQ de manière
« quasi-électronique » par mise à
disposition sur le réseau
informatique avec une indication de la date de mise à jour de la
liste.
Les
documents techniques
liés au produit sont gérés par les responsables
concernés (Conception,
Production, Achats).
Pour la
conception, des
répertoires informatiques permettent le classement des
documents et des
enregistrements de la conception, avec une architecture sur le
Réseau
informatique par projet de conception et selon des
répertoires types, par
exemple:
·
Planification
(Plan de développement, planning, Jalonnement,….)
·
Données
d’entrée
(Besoins du marché, Spécifications Marketing,…)
·
Données
de Sortie
(Spécifications technique, Architecture,
Plans, Nomenclature, Rapport de
gestion de risques …)
·
Revues
de Conception
(CR de revue de projet et/ou de conception, CR de passage de Jalons)
·
Vérification
(Plans de tests, rapports de tests,…)
·
Validation
(Données cliniques, Protocole et rapport d’évaluation
externe par des
utilisateurs,…)
·
Dossiers
Réglementaires
(Index de la documentation technique marquage CE, Index du D.H.F Design
History
File, D.M.R. Index du Device Master Record)
Par
ailleurs, lors des
revues de conception, les documents pertinents revus sont
compilés dans un
classeur papier à chaque étape de conception.
Le
réseau informatique est
sauvegardé périodiquement sur un média externe.
Préalablement
à la revue de
direction, le RQ liste tous les documents revus et approuvés
depuis plus de 3
ans afin de déterminer s’il convient de mettre à jour
certains de ces documents
et proposer un plan d’actions en ce sens à la revue de direction.
Les
enregistrements sont
établis et conservés pour apporter la preuve de la
conformité aux exigences et
du fonctionnement efficace du système de management de la
qualité.
Ces
enregistrements sont
conçus, établis et conservés de manière
à rester lisibles, faciles à identifier
et accessibles.
Les
modalités pour
l'identification, le stockage, la protection, l'accessibilité,
la durée de
conservation et l'élimination des enregistrements sont
indiquées dans la Liste
L02 Liste des Enregistrements.
Les
modalités pour déterminer la durée de conservation
des enregistrements sont
définies comme ci-dessous :
·
Pour
les enregistrements du SMQ (QSR), une durée suffisante pour
démontrer le
fonctionnement du SMQ (au moins 3 à 4 périodes de
renouvellement)
·
Pour
les documents techniques du SMQ liés aux dispositifs
médicaux (D.M.R, D.H.R,
D.H.F et Documentation Technique marquage CE) pendant une durée
au moins
équivalente à la durée de vie du dispositif
médical.
La
durée de vie est définie de la manière
suivante : durée pendant laquelle
il existe au moins un dispositif médical en cours d’utilisation
dans la base
installée.
La
Base installée est gérée via les opérations
de livraison, d’installation, et de
maintenance. De plus, les obligations liées au décret
2005-829 implique un
suivi par le « Producteur » des déchets
des D.E.E.E (Déchets des
Equipements Electriques et Electroniques) pour la collecte et la
valorisation
de ces déchets.
L’ensemble
des documents techniques et des enregistrements Qualité
concernant le produit
(plans, nomenclatures, instructions de travail, résultats de
contrôle, ...) est
conservé pendant la durée de vie des produits (avec un
minimum de 5 ans après
avoir obtenu l’assurance que la base installée ne comporte plus
aucun produit
en service).
ANNEXE 3 : SOMMAIRE
DU MANUEL QUALITE
0- INTRODUCTION
0.1
Généralités
1- DOMAINE
D’APPLICATION
1.1
Généralités
Savoir faire
Secteurs
d’activité
Renseignements
1.2
Application
2- REFERENCES
NORMATIVES
3- TERMES ET
DEFINITIONS
4- SYSTEME DE
MANAGEMENT DE LA QUALITE
4.1
Exigences générales
4.2
Exigences relatives à la documentation
5- RESPONSABILITE DE
LA
DIRECTION
5.1
Engagement de la direction
5.2
Ecoute client
5.3
Politique Qualité
5.4
Planification
5.4.1 Objectifs qualité
5.4.2 Planification du système de
management
de la qualité
5.5
Responsabilité, autorité et communication
5.6
Revue de direction
6- MANAGEMENT DES
RESSOURCES
6.1
Mise à disposition des ressources
6.2
Ressources humainesErreur !
Signet non défini.
6.3
Infrastructures
6.4
Environnement de travail
7- REALISATION DU
PRODUIT
7.1
Planification de la réalisation du produit
7.2
Processus relatifs aux clients
7.3
Conception et développement
7.4
AchatsErreur !
Signet non défini.
7.5
Production et préparation du service
7.6
Maîtrise des dispositifs de surveillance et
de mesure
8- MESURE, ANALSE ET
AMELIORATION
8.1
Généralités
8.2
Surveillance et mesures
8.3
Analyse des données
8.4
Amélioration
9- VEILLE
REGLEMENTAIRE
ET NORMATIVE
retour sommaire
.jpg)