Master Qualité - Communication
publique des résultats d'un stage de fin d'études UTC - rue Roger Couttolenc - CS
60319 - 60203 Compiègne Cedex - France - master-qualite@utc.fr
- Tél : +33 (0)3 44 23 44 23
Avertissement
: Si vous arrivez directement sur cette page, sachez
que ce travail est un rapport d'étudiants et doit être
pris comme tel. Il peut donc comporter des
imperfections ou des imprécisions que le lecteur doit
admettre et donc supporter. Il a été réalisé pendant
la période de formation et constitue avant-tout un
travail de compilation bibliographique, d'initiation
et d'analyse sur des thématiques associées aux
concepts, méthodes, outils et expériences sur les
démarches qualité dans les organisations. Nous ne
faisons aucun usage commercial et la duplication est
libre. Si, malgré nos
précautions, vous avez des raisons de contester ce
droit d'usage, merci de nous
en faire part, nous nous efforcerons d'y
apporter une réponse rapide. L'objectif de la
présentation sur le Web est de permettre l'accès à
l'information et d'augmenter ainsi les échanges
professionnels. En cas d'usage du document, n'oubliez
pas de le citer comme source bibliographique.
Bonne lecture...
Référentiels
qualité majeurs pour les entreprises biomédicales :
outil d'auto-diagnostic
Référence
bibliographique à rappeler pour tout usage : Référentiels qualité majeurs pour les entreprises biomédicales : outil d'auto-diagnostic, EL
MARSAOUI Hamza, LAMKADEM Wiame, MANCET Claire, MEKSI
Lamjed Université de
Technologie de Compiègne, Master Qualité et
Performance dans les Organisations (QPO) ou Master
Technologies et Territoires de Santé (TTS),
Mémoire d'Intelligence Méthodologique du projet
d'intégration, janvier 2018, www.utc.fr/master-qualite,
puis "Travaux", "Qualité-Management", réf n° 42, https://doi.org/10.34746/k7n3-sb47
RESUME
Ce Mémoire d’Intelligence Méthodologique décrit le
projet d’un groupe d’étudiants des Masters Technologies
et Territoires de Santé et Qualité et Performance dans
les Organisations de l’Université Technologique de
Compiègne (France).
L’objectif des travaux consiste à fournir aux
entreprises biomédicales un outil d’autodiagnostic pour
mesurer et améliorer leur système qualité et les
accompagner, le cas échéant, dans une démarche de
certification afin d’accélérer la mise sur le marché des
dispositifs médicaux. L’outil d’autodiagnostic repose
sur trois normes fondamentales pour ce type
d’entreprises : ISO 9001 : 2015, ISO 13485 : 2016, ISO
14971 : 2013.
Mots clés : Entreprise biomédicale,
innovation, enjeux de compétitivité, risques,
sécurité, ISO 9001, ISO13485, ISO 14971, outil
d’autodiagnostic.
ABSTRACT
This Methodological
Intelligence Memory deals with a project led by a group
of students of the University of Technology of Compiègne
(France) (Masters in Technologies and Territories of
Health and Quality and Performance in Organization).
The scope of this work aims at providing biomedical
companies with a self-diagnostic tool based on three
fundamental standards for such a kind of companies: ISO
9001: 2015, ISO 13485: 2016, ISO 14971: 2013.
Keywords: Biomedical company, innovation,
competitiveness concerns, risks, safety, ISO 9001,
ISO13485, ISO 14971, self-assessment tool.
Nous tenons à remercier Monsieur Jean-Matthieu PROT
Co-Responsable du Master Technologie et Territoires de Santé et
Monsieur Gilbert FARGES, Responsable du Master Qualité et
Performance dans les Organisations de l’UTC, pour leurs précieux
conseils et leur accompagnement dans les divers jalons de notre
projet.
▪ Cycle de vie du Dispositif Médical :
Toutes les phases de la vie d’un dispositif médical, depuis sa
conception initiale jusqu’à sa mise hors service et sa mise au rebut
finales.[1]
▪ Dispositif médical [2]:
Tout instrument, appareil, équipement, logiciel, implant, réactif,
matière ou autre article, destiné par le fabricant à être utilisé,
seul ou en association, chez l'homme pour l'une ou plusieurs des
fins médicales précises suivantes:
— diagnostic, prévention, contrôle, prédiction, pronostic,
traitement ou atténuation d'une maladie, diagnostic, contrôle,
traitement, atténuation d'une blessure ou d'un handicap ou
compensation de ceux-ci,
— investigation, remplacement ou modification d'une structure ou
fonction anatomique ou d'un processus ou état physiologique ou
pathologique,
— communication d'informations au moyen d'un examen in vitro
d'échantillons
provenant du corps humain, y compris les dons d'organes, de sang et
de tissus,
et dont l'action principale voulue dans ou sur le corps humain n'est
pas obtenue par des moyens pharmacologiques ou immunologiques ni par
métabolisme,
mais dont la fonction peut être assistée par de tels moyens.
▪ Dispositifs médicaux logiciels et applications
mobiles en santé :
DM ou DMIV destiné à une utilisation à des fins médicales.
Il permet un diagnostic, une aide au diagnostic, un traitement ou
une aide au traitement du patient. Il fournit une information
médicale nouvelle contribuant par exemple au diagnostic ou au
traitement du patient.[3]
▪ Entreprise biomédicale:
Toute entreprise (ETI, PME, TPE, start-up …) développant et
commercialisant des dispositifs médicaux innovants (dispositif
médicaux et dispositifs de diagnostic in vitro d’innovation parmi
lesquels on trouvera notamment des logiciels et applications mobiles
en santé).
▪ Fabricant:
Personne physique ou morale responsable de la conception et/ou de la
fabrication d’un dispositif médical dans le but de le rendre
disponible pour utilisation, en son nom, que ce dispositif médical
soit ou non conçu et/ou fabriqué par cette personne ou en son nom
par une ou plusieurs autres personnes.
▪ Norme:
La norme est un document élaboré par consensus au sein d’un
organisme de normalisation par sollicitation des représentants de
toutes les parties intéressées. Son adoption est précédée d’une
enquête publique.
▪ Outil d’autodiagnostic :
Grille utilisée pour mesurer une activité complexe selon plusieurs
dimensions.[4]
DM : Dispositif Médical
DMDIV : Dispositif Médical de Diagnostic In Vitro
EBI : Entreprise Biomédicale Innovante
ETI : Entreprise de Taille Intermédiaire
PME : Petites et Moyennes Entreprises
PMS : Post Market Surveillance
TPE : Très Petite Entreprise
TTS : Technologies et Territoires de Santé
QPO : Qualité et Performance dans les Organisations
R&D : Recherche et Développement
SCAC : Suivi Clinique après Commercialisation
SPAC : Suivi des performances après commercialisation
UDI : Identification unique du Dispositif Médical
Liste des figures
Figure 1 : Chiffre d’Affaires réalisé sur le marché biomédical selon
la typologie de DM
Figure 2 : Cycle de vie d’un DM (Directives Nouvelle Approche)
Figure 3 : Cycle de vie d’un DM (Nouveau Règlement Européen DM)
Figure 4: Cartographie de la norme ISO 9001 : 2015
Figure 5 : Cartographie de la norme ISO 13485 :2016
Figure 6 : Représentation schématique du processus de gestion des
risques
Figure 7 : Cartographie de la norme ISO 14971 :2013
Figure 8 : Démarche de mutualisation des normes
Figure 9 : Etude comparative des trois normes selon les chapitres
ISO 13485
Figure 10: Etude comparative des trois normes par thème
Figure 11 : Avantages et inconvénients des types d’outils et de
leurs supports
Figure 12 : onglet du mode d’emploi de l’outil
Figure 13: onglet d’évaluation de l’outil d’auto-diagnostic.
Figure 14 : Tableau des couleurs associées à chaque exigence.
Figure 15 : Tableau des niveaux de véracité
Figure 16: Tableau des niveaux de conformité par chapitre
Figure 17: Présentation des résultats par articles et sous-articles
Figure 18 : Tableau de synthèse des résultats
Figure 19: Présentation des résultats de conformité pour chaque
norme
Figure 20: Conseils pour atteindre le niveau de conformité
Figure 21 : Logigramme de synthèse des apports de l’outil
d’autodiagnostic
Les entreprises biomédicales doivent assurer la qualité des
dispositifs médicaux qu’elles souhaitent mettre sur le marché, tout
en respectant les exigences réglementaires pour garantir la sécurité
des patients, en optimisant les temps de développement et de mise
sur le marché des produits afin d’être compétitives et de préserver
leur image de marque.
Elles doivent obtenir un marquage CE et donc mettre en place un
système qualité efficace et efficient et répondant aux exigences
réglementaires et normatives selon leur secteur d’activités, ce qui
s’avère être un exercice complexe.
Les travaux menés ont donc eu pour objectif de:
Évaluer le contexte et les enjeux des entreprises
biomédicales
Fournir à ces entreprises un outil pour mesurer et
améliorer leur système qualité et les accompagner, le cas
échéant, dans une démarche de certification (dans l’optique
d’accélérer la mise sur le marché des dispositifs médicaux et
répondre ainsi plus rapidement aux besoins médicaux non
satisfaits).
Ce Mémoire d’Intelligence Méthodologique relate donc la démarche
d’élaboration d’un outil de positionnement (ou outil
d’auto-diagnostic) "3-DIAG" sur 3 référentiels d’importance pour
l'activité de toute entreprise biomédicale:
✓ ISO 9001 : 2015 Système de Management de la
Qualité-Exigences [5]
✓ ISO 13485 Dispositifs médicaux Systèmes de
management de la qualité Exigences à des fins réglementaires-avril
2016 [6]
✓ ISO 14971 : 2013 Dispositifs médicaux -
Application de la gestion des risques aux dispositifs médicaux [7]
Le présent document décrit les entreprises biomédicales, leur
contexte socio-économique et réglementaire ainsi que les enjeux
actuels de la filière biomédicale.
Il aborde en outre les objectifs des travaux et les critères rendant
incontournables les normes précitées.
Il détaille, pour finir, l’approche méthodologique qui a été choisie
pour élaborer l’outil d’auto-diagnostic "3-DIAG" ainsi que son
exploitation.
I.
Les entreprises biomédicales : normes et enjeux
1.
Le contexte des entreprises biomédicales
Les entreprise biomédicales conçoivent, fabriquent et/ ou
commercialisent des DM qui doivent répondre aux exigences des
professionnels de santé et aux besoins des patients.
Pour cela, elles investissent dans la R&D dans le domaine de
santé et de la création de produits, procédés ou techniques qui
répondent à des enjeux de santé publique majeurs.
D’après une étude du SNITEM [8], le secteur des dispositifs médicaux
en France se caractérise par :
▪ 1 343 entreprises recensées qui réalisent un
chiffre d’affaires de 28 Mds d’euros avec une très forte dominante
de PME (92 %).
▪ 4% de croissance par an en moyenne
▪ 85.000 emplois
▪ Plus de la moitié des entreprises ont une
activité de R&D, près de 13% sont exclusivement actives en
R&D (start-up), près de 60% ont une activité de production et
plus de 80% une activité commerciale
Plus précisément la filière d’entreprises biomédicales regroupe :
▪ 214 entreprises nouvellement créées (comprenant
une large majorité de start-up)
▪ 411 entreprises nouvellement visibles
(entreprises préexistantes mais qui n’avaient pas positionné leurs
activités dans le domaine du dispositif médical.
▪ 361 entreprises ayant fait l’objet de rachats,
de fusions, de repositionnement ou de liquidations.
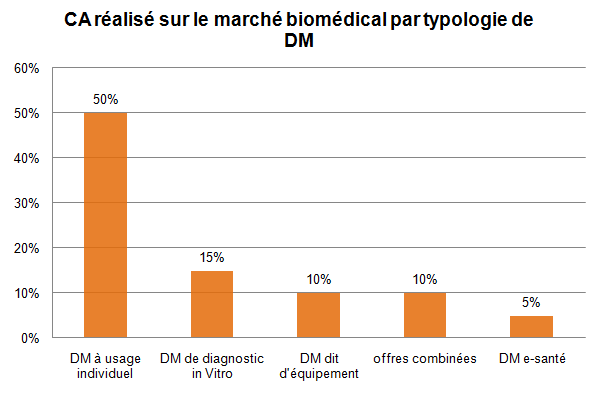
Figure 1 : Chiffre d’Affaires réalisé sur le marché biomédical
selon la typologie de DM [5]
La filière biomédicale enregistre une croissance annuelle du secteur
à l’export de 5%.
Le SNITEM précise dans sa conclusion de l’étude que « l’innovation
reste un enjeu incontournable pour les entreprises » et qu’ «elle
s’illustre par une volonté d’élaborer des offres de solutions
intégrant de nouvelles fonctionnalités, issues notamment des
technologies de e-santé ».
En 2015, on comptait 14445 brevets avec une majorité détenue par des
entreprises américaines. (Source : QUESTEL).
Les entreprises biomédicales commercialisent plusieurs types de DM :
• Les DM à usage unique,
• Les DM de diagnostic in vitro,
• Les DM implantables actifs,
• Les équipements médicaux.
•
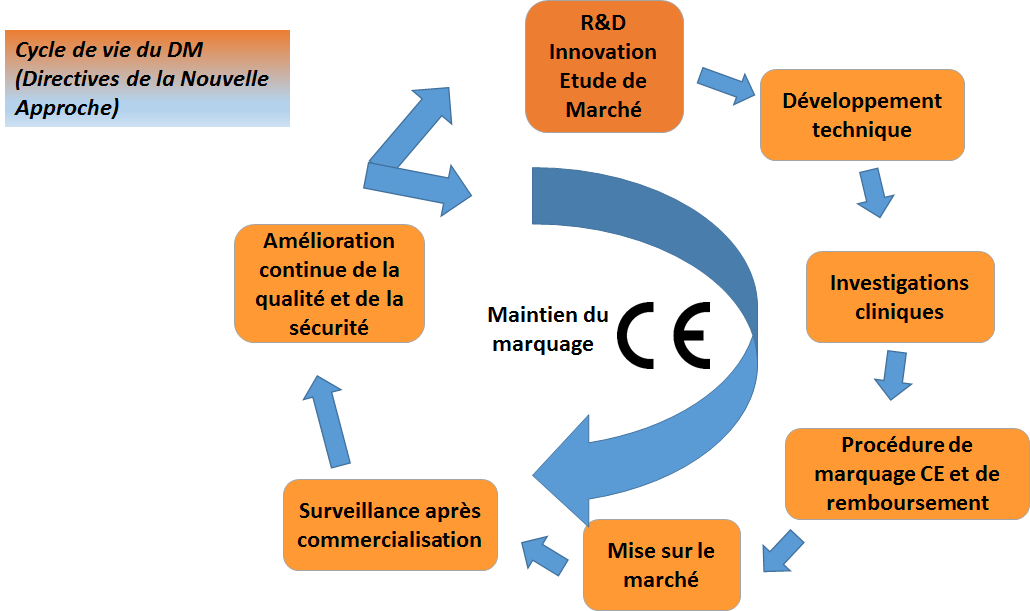
Le cycle de vie d’un DM comprend les étapes suivantes :
Figure 2 : Cycle de vie d’un DM (Source : Auteurs)
En Europe, les fabricants doivent démontrer la conformité aux
exigences essentielles pour obtenir un marquage CE et donc pour
mettre leurs DM sur le marché, conformément aux Directives de la
Nouvelle Approche 98/79/CE du 27/10/1998 (DM de diagnostic in
vitro), 90/385/CEE du 20/06/1990 (DM implantables actifs) et
93/42/CEE du 14/06/1993 (Dispositif médicaux).
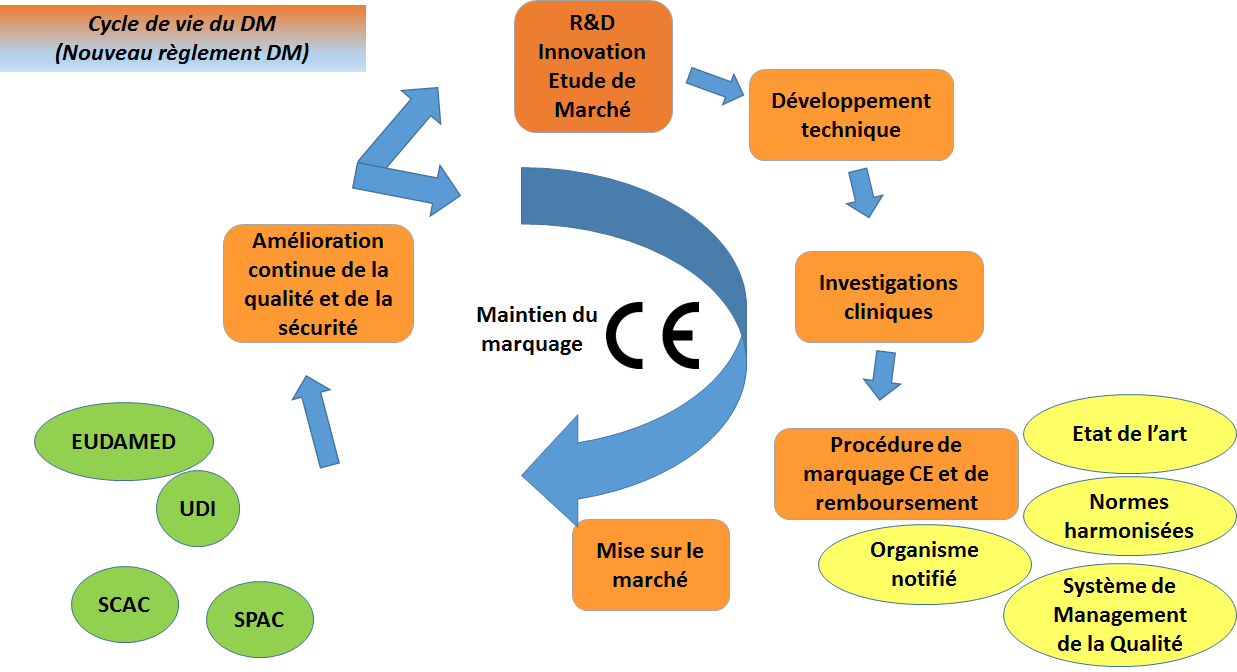
Un nouveau règlement européen sur les dispositifs médicaux [3] et
sur les dispositifs médicaux in vitro [10] a été adopté en avril
2017 pour être en phase avec les innovations technologiques telles
que les logiciels et nanomatériaux, pour garantir la sécurité des
patients et pour harmoniser les exigences en termes d’évaluation
clinique et de matériovigilance dans les différents pays de l’Union.
Ce règlement renforce les exigences essentielles et entrera en
vigueur en 2020. Il remplacera donc les Directives.
Les principaux changements introduits par le Règlement sont les
suivants :
Définition élargie du Dispositif Médical (introduction de la
notion d’implant et de réactif)
Durcissement des règles de classification du DM
Procédure d’évaluation de conformité renforcée selon la classe
du DM (SMQ obligatoire sauf pour les DM de classe I)
Exigences essentielles renforcées
Documentation technique renforcée (pour les aspects cliniques
et PMS)
Rôles et obligations définis plus précisément pour le
fabricant, importateur, distributeur, mandataire.
Introduction d’une personne chargée de veiller au respect de
la réglementation
Renforcement de la surveillance par les organismes notifiés
Renforcement des exigences pour les évaluations cliniques et
pour la surveillance après commercialisation (SPAC, SCAC, suivi
des incidents)
Création de la base de données EUDAMED accessible aux
autorités pour suivre les enregistrements, les investigations
cliniques, la matériovigilance et la surveillance du marché.
Suite au nouveau règlement, les fabricants doivent vérifier la
classe de leurs DM sur le marché ou en développement et mettre à
niveau leurs systèmes de management de la qualité et de gestion des
risques [2] sur les dispositifs médicaux qu'ils conçoivent,
développent, industrialisent, maintiennent et suivent sur le marché.
Plus précisément, le fabricant doit vérifier que les produits sont
conformes à leur usage, garantir la sécurité du patient et des
utilisateurs, supprimer, réduire ou atténuer les risques identifiés
et communiquer sur les risques résiduels. Il devra in fine démontrer
que les risques résiduels sont acceptables en comparaison des
bénéfices pour le patient. Pour cela le fabricant doit s’appuyer sur
les normes existantes et sur l’état de l’art concernant le système
de management de la qualité, la sécurité et la compatibilité.
La classe de risque d'un dispositif médical est définie selon sa
dangerosité potentielle pour le patient. Le fabricant peut s'appuyer
sur 22 règles et 80 critères figurant sur le nouveau règlement
européen (contre 18 règles figurant sur la Directive 93/42/CEE) pour
l'identification de la classe de son dispositif médical. Les
dispositifs médicaux de classe I ne requièrent pas obligatoirement
l'intervention d'un organisme notifié pour l'obtention de marquage
CE. En revanche, pour les autres classes (IIa, IIb et III), les
recours à un organisme notifié est obligatoire. Quelle que soit la
classe du dispositif, les fabricants ont avantage à se conformer à
l'ISO 9001 et l'ISO 13485 qui sont les deux principales normes
relatives aux systèmes de management de la qualité et à la norme ISO
14971 qui décrit la méthodologie d'analyse de risque appliquée aux
dispositifs médicaux.
La conformité aux normes harmonisées publiées au Journal Officiel de
l’Union Européenne vaut présomption de conformité aux exigences
essentielles du nouveau règlement européen. La norme ISO 13485 :
2012 et la norme ISO 14971 : 2012 figurent dans la liste des normes
harmonisées et la norme ISO 13485 : 2016 est applicable et sera
prochainement harmonisée.
Figure 3 : Cycle de vie d’un DM avec le
nouveau règlement DM (Source : Auteurs)
Aux États-Unis, les fabricants de dispositifs médicaux
doivent mettre en œuvre et suivre un système qualité conforme
aux Bonnes Pratiques de Fabrication (Good Manufacturing
Practice) qui incluent depuis 1987 les exigences de la norme ISO
9001 et de la norme ISO 13485 [11].
Au Japon, depuis 2014, l’ordonnance ministérielle portant sur
la fabrication et le contrôle qualité des dispositifs médicaux
et des dispositifs médicaux de diagnostic in vitro a été révisée
et harmonisée avec la norme ISO 13485 :2003. Les responsables de
la mise sur le marché de dispositifs médicaux doivent s’assurer
que le site de fabrication respecte les normes, conserve les
documents et les enregistrements conformément aux durées
réglementairement fixées et dispose d’un système de transmission
des incidents [12].
Compte-tenu des spécificités et de la répartition des activités des
entreprises biomédicales, de leurs enjeux, des principales zones de
commercialisation et des exigences réglementaires internationales,
il a paru pertinent de privilégier trois normes en particulier pour
:
ISO 9001 : 2015 : Systèmes de management de la
Qualité-Exigences
ISO 13485 Dispositifs médicaux Systèmes de management de la
qualité- Exigences à des fins réglementaires-avril 2016
ISO 14971 Dispositifs médicaux - Application de la gestion des
risques aux dispositifs médicaux- janvier 2013
2. ISO 9001 : 2015 Systèmes
de management de la qualité — Exigences
Un grand nombre d’entreprises choisissent cette norme car elle vise
à fournir en continu des produits et des services conformes aux
attentes des clients et aux exigences réglementaires et légales.
Ainsi, l’ISO 9001 ressort comme la certification la plus délivrée
selon l’enquête 2015 d’ISO Survey, avec plus d’un million de
détenteurs dans le monde. En France, 27 844 structures détenaient ce
certificat de management de la qualité fin 2015 [13].
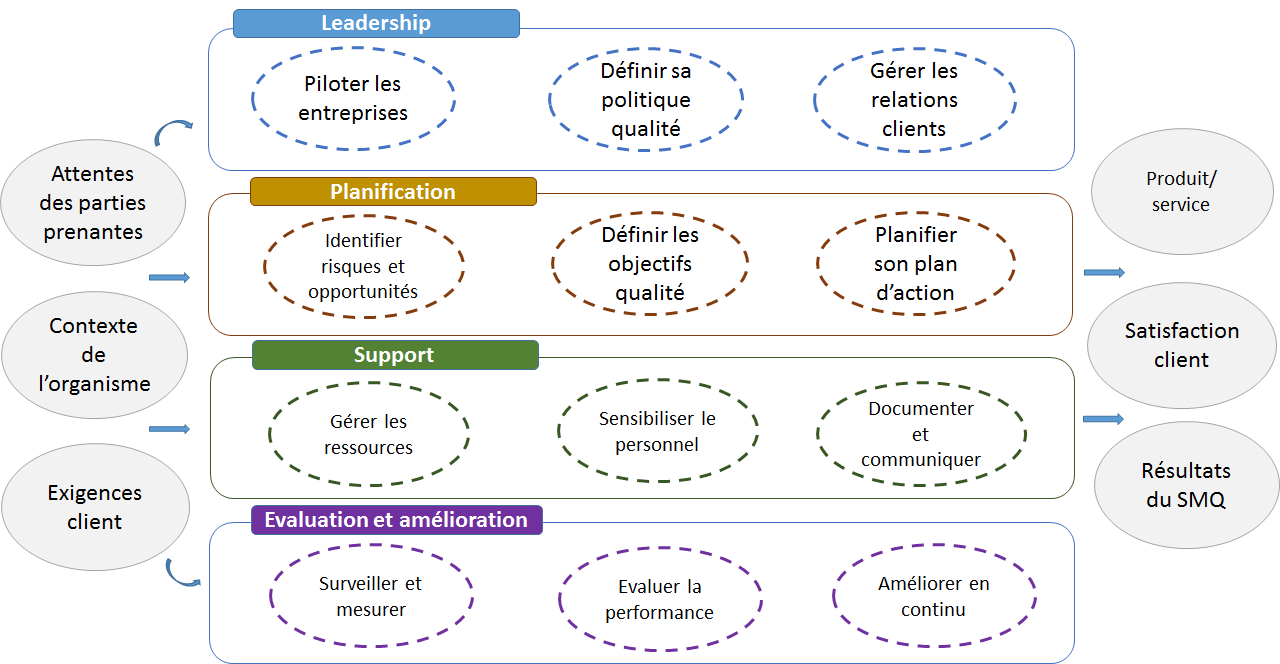
Cette norme explicite les exigences pour la mise en place d’un
système de management de (par) la qualité :
Le contexte de l’organisme : la compréhension de l’organisme
et de son contexte, la compréhension des besoins et des attentes
des parties intéressées, la détermination du domaine
d’application du SMQ, le système de management de la qualité et
les processus associés.
La planification : les actions à mettre en œuvre face aux
risques et aux opportunités, les objectifs qualité et la
planification pour les atteindre, la planification des
modifications.
Le leadership : leadership et engagement, politique, rôles,
responsabilités et autorités au sein de l’organisme.
Le support : ressources, compétences, sensibilisation,
communication, informations documentées
La réalisation des activités opérationnelles : planification
et maîtrise opérationnelles, exigences relatives aux produits et
services, conception et développement des produits et services,
maîtrise des processus, produits et services fournis par des
prestataires externes, production et prestation de services,
libération des produits et services, maîtrise des éléments de
sortie non conforme.
L’évaluation des performances : surveillance, mesure, analyse
et évaluation, audit interne, revue de direction.
L’amélioration : non-conformité et actions correctives,
amélioration continue.
3. ISO 13485 Dispositifs
médicaux Systèmes de management de la qualité Exigences à des
fins réglementaires-avril 2016
Cette norme (constituée de 8 chapitres et 5 annexes) s’adresse aux
entreprises impliquées dans les étapes du cycle de vie d’un
dispositif médical ainsi qu’à leurs fournisseurs ou tierces parties
et vise à la sécurité des patients, à satisfaire aux exigences des
clients et aux exigences réglementaires, tout en incluant la gestion
des risques.
Elle détaille les exigences :
• réglementaires,
• relatives à la conception, au développement et à
l’industrialisation du dispositif médical,
• relatives à la validation et à la documentation.
La gestion des risques tient une part importante en termes de
maîtrise de processus, des fournisseurs, de validation des logiciels
et maîtrise des non-conformités des dispositifs médicaux.
La période de transition pour la certification selon cette version
court jusqu’à février 2019.
Durant cette période, les entreprises doivent donc étudier la norme
et réaliser un bilan des écarts de leurs SMQ actuel par rapport aux
nouvelles exigences de la norme. L’outil d’auto-diagnostic est donc
un atout pour cette étape.
Les exigences de la norme en termes de documentation sont
importantes :
- 1 dossier de conception du DM
- 24 documents
- 30 procédures
- 16 enregistrements
L’annexe A de la norme décrit les modifications apportées à la
version 2016 par rapport à la version précédente de la norme
(version 2003).
L’annexe B donne deux tableaux de correspondances entre les articles
et paragraphes de l’ISO 13485 : 2016 et l’ISO 9001 :2015.
Les annexes ZZA, ZZB ZZC détaille relations avec les Directives
Européennes et les exigences couvertes ou non par la norme.
Figure 5 : Cartographie de la norme ISO 13485 :2016 (Source :
Auteurs)
4. ISO 14971 Dispositifs
médicaux - Application de la gestion des risques aux
dispositifs médicaux- janvier 2013
Cette norme de 9 chapitres (et 13 annexes) s’adresse aux fabricants
de dispositifs médicaux et vise à d'identifier les phénomènes et
situations dangereuses associés aux DM ET DMDIV, à estimer et
évaluer les risques, à maîtriser ces risques et à surveiller
l'efficacité de cette maîtrise.
L’analyse des risques consiste à vérifier l’emploi prévu et à
identifier les caractéristiques associées à la sécurité du DM,
identifier les phénomènes dangereux et à estimer les risques pour
chaque situation dangereuse.
La maîtrise du risque comprend la mise en œuvre des mesures,
l’évaluation des risques résiduels et les risques découlant
des mesures.
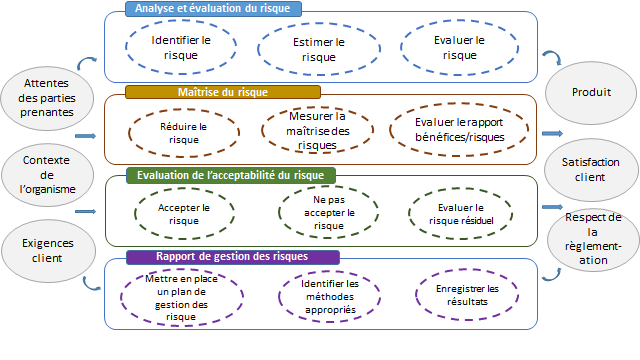
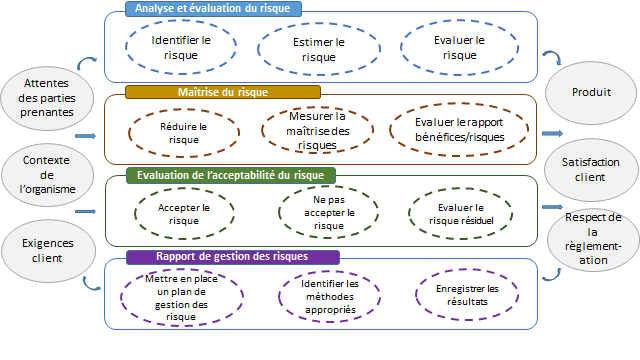
La norme comprend un schéma descriptif résumant le processus de
gestion des risques :
Figure 6 : Représentation schématique du
processus de gestion du risque [7]
La norme décrit également l’importance de l’implication de la
Direction dans le processus de Gestion des Risques par le maintien
des ressources adéquates et des revues périodiques.
La Direction doit également s’assurer que le personnel chargé
d’effectuer les tâches de gestion des risques est qualifié et
veiller à la mise en place d’un plan de gestion des risques et d’un
dossier de gestion des risques pour le DM.
Le Plan de Gestion des Risques fait partie du Dossier de Gestion des
Risques et permet d’identifier le domaine d’application du plan
selon le DM et la phase du cycle de vie du DM concernée,
l’attribution des responsabilités, la revue des activités , les
critères d’acceptabilité des risques, la vérification et les
activités de collecte et revue des informations de production et de
post-production.
En établissant un rapport de gestion des risques, le fabricant
apporte la preuve de mise en œuvre du plan de gestion des risques et
que les objectifs ont été atteints.
L’annexe A de la norme présente une justification des différentes
exigences.
L’annexe B donne un schéma récapitulatif du processus de gestion des
risques.
L’annexe C décrit les questions que le fabricant doit se poser pour
identifier les caractéristiques des DM.
L’annexe D décrit les concepts de risques appliqués aux DM.
L’annexe E cite des exemples de phénomènes dangereux, séquences
d’événements prévisibles et situations dangereuses.
L’annexe F décrit le contenu du Plan de Gestion des Risques.
L’annexe G décrit les informations sur les techniques de gestion des
risques.
L’annexe H comprend les lignes Directrices sur la gestion des
risques liées aux DM in vitro.
L’annexe I décrit les lignes directrices relatives aux processus
d’analyse des phénomènes dangereux.
L’annexe J décrit les informations relatives à la sécurité et au
risque résiduel.
Les trois dernières annexes ZZA, ZZB et ZZC établissent les
relations entre la norme et les Directives de la Nouvelle Approche.
Figure 7: Cartographie de la norme ISO 14971 :2013 (Source
: Auteurs)
5. Les
enjeux des entreprises biomédicales et les
leviers de la certification
Enjeux techniques
La mission d’une entreprise biomédicale est orientée par le progrès
technique, scientifique en concertation avec les professionnels de
santé (hôpitaux, ingénieurs biomédicaux, médecins, chercheurs, …)
pour répondre aux besoins médicaux de la population.
Pour répondre à ce besoin, une entreprise biomédicale doit :
• faire des études de marché afin d’évaluer la
faisabilité technique du produit, d’écouter le besoin des clients
(professionnels de santé, patients) et d’identifier les moyens/
ressources nécessaires.
• proposer des solutions techniques et innovantes
de réalisation du produit afin de se démarquer de la
concurrence.
• répondre à un ensemble d’exigences
réglementaires et normatives du domaine médical (classification du
DM, obtention du marquage CE, certifications etc…);
• offrir aux acteurs de santé des dispositifs
médicaux de haute technologie qui permettent de faciliter et suivre
le traitement du patient et le diagnostic médical.
Les enjeux sociétaux
Ils consistent à augmenter la motivation de leur personnel et
assurer une veille technologique pour développer des dispositifs
médicaux à la pointe de l’innovation et facile d’utilisation,
encourager la créativité et instaurer une culture d’initiative.
Les enjeux financiers
L’entreprise doit :
• s’adapter aux difficultés d’investissement, la
recherche d’un fond de roulement restant le premier défi pour les
entreprises biomédicales.
• équilibrer son budget d’investissement sur les
compétences techniques de leurs personnels, le cout, la qualité et
le délai de la fabrication/commercialisation de leurs dispositifs
médicaux ;
• tenir compte des coûts inhérents au processus de
certification aux normes (incluant des coûts directs tels que les
coûts liés aux frais de dossier de l’organisme certificateur et à
l’audit de certification et des coûts indirects liés à la gestion du
personnel, à la formation, à la mise œuvre d’un plan d’action
qualité.
• mettre en place des essais cliniques
indispensables pour démontrer la conformité des dispositifs médicaux
aux exigences essentielles de la directive 93/42/CEE (dispositifs
médicaux), et la directive 90/385/CEE (dispositifs médicaux
implantables actifs) [14].
Les normes sont des alliées pour faire face à ces enjeux car elles
fournissent aux entreprises un support pour développer leurs
systèmes de management de la qualité et de management du risque.
Le projet d’outil d’autodiagnostic mutualisé selon les trois normes
ISO 9001:2015, ISO 13485: 2016 et ISO 14971:2013 permet aux
entreprises de réduire leurs coûts de certification, puisqu’une
seule procédure de certification pourra être suivie pour les trois
normes simultanément.
La triple certification permettra aux entreprises biomédicales
d’impliquer et de motiver leur personnel, d’améliorer leur image, de
maîtriser leurs processus, d’obtenir et maintenir leurs marquages CE
et donc de vendre leurs produits, d’améliorer la qualité de leurs
produits et de leurs services et d’assurer la sécurité des patients
et des utilisateurs des dispositifs médicaux.
II. Etude, mutualisation
des exigences normatives et conception de l’outil
d’autodiagnostic "3-DIAG"
1. Mutualisation des
exigences
Pour cette étape, la réflexion a été basée sur les travaux d’un
groupe d’étudiants datés de 2016, portant sur « La nouvelle ISO 9001
(2015) et future ISO 13485 (2016) : Mutualisation des exigences et
outil bi-diagnostic pour la performance des entreprises biomédicales
» [4].
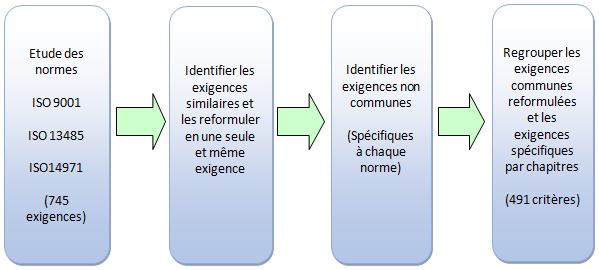
Les travaux consistent à analyser les exigences similaires entre les
trois normes selon la démarche suivante :
Figure 8: Démarche de mutualisation des
normes (Source : Auteurs)
A l’issue du travail de mutualisation des exigences, il est
nécessaire de reformuler les exigences communes aux trois normes et
celles spécifiques à chacune d’entre elles en phrases simples et
compréhensibles par toute personne non familiarisée à la sémantique
de la qualité.
A partir des 745 exigences des trois normes, le processus de
mutualisation permet d'évaluer seulement 491 critères dont 227
spécifiques et 264 communs.
Etude comparative des normes
L’étude comparative des normes a été faite chapitre par chapitre. La
comparaison a été faite également en se basant sur les chapitres de
la norme ISO 13485 et en s’appuyant sur son annexe B.
Chapitres
de la norme ISO 13485 :2016
Exigences
ISO 13485 :2016
ISO 9001
ISO
14971 :2013
Système
de Management de la Qualité
Contexte
réglementaire
Politique
Qualité
Manuel
Qualité
Documentation
de validation
Procédures
Dossier
DM
Maîtrise
des documents
Maîtrise
des enregistrements
Contexte
de l’organisme
Vérification
du processus de gestion des risques à intervalles
définis
Responsabilité
de la Direction
(Reformulé
en Gestion des responsabilités)
Engagement
de la Direction
Orientation
client
Politique
Qualité,
Planification,
Responsabilité,
autorité, communication, revue de direction
Leadership
Responsabilité
de la Direction dans la Gestion du Risque
Politique
de Gestion du Risque
Management
des ressources
Mise à
disposition des ressources,
Ressources
humaines
Infrastructures,
environnement de travail et maîtrise de la contamination
Ressources
Ressources
humaines
Compétences
Sensibilisation
Infrastructure
Environnement
pour la mise en œuvre des processus.
Ressources
Humaines : qualification du personnel
Ressources
adéquates pour la gestion des risques
Réalisation
du produit
Planification
de la réalisation du produit
Exigences
clients, exigences produits
Conception
et développement, (planification, éléments d’entrée et
de sortie, revue, vérification, validation, transfert,
maîtrise), achats, production et prestation de service,
identification, traçabilité
Réalisation
des activités opérationnelles
Mesurage,
analyse et amélioration
Retour
d’information,
Traitement
des réclamations,
Signalement
aux autorités réglementaires, audit interne,
surveillance et mesure des processus et des produits,
maîtrise des non-conformités, amélioration
Evaluation
des performances
Analyse
du risque
Analyse
des informations de production et de postproduction
Figure 9: Etude comparative des 3 normes
selon les chapitres de l’ISO 13485 :2016 (Source : Auteurs)
L’outil d’autodiagnostic développé correspond donc à un fichier
Excel
®
contenant des formules de calcul simples, facile d’accès et
d’utilisation.
Les chapitres des exigences sont basés sur la norme ISO 9001 : 2015
car cette norme décrit le système de management de la qualité propre
aux dispositifs médicaux et donc aux entreprises biomédicales.
3. Structure de l’outil
"3-DIAG"
L’outil proposé est un fichier Excel® automatisé, simple autoporteur de
sens, fonctionnel et ergonomique.
Il s’agit d’une solution efficace pour mesurer la conformité du SMQ
aux exigences des normes et identifier les axes prioritaires
d’amélioration, en une durée de 2 heures et 30 minutes maximum. (491
critères).
L’outil est constitué de 10 onglets :
{Menu} : Interface de navigation où l'utilisateur peut
sélectionner l'onglet désiré.
{Page d'accueil} : Onglet interactif dans lequel
l'utilisateur peut trouver les instructions d'utilisation de l'outil
et les échelles d'évaluation :
Échelle de véracité à 6 niveaux pour évaluer les critères :
Non-concerné, Faux unanime (0%), Faux (20%), Plutôt faux (40%),
Plutôt vrai (60%), Vrai (80%), Vrai prouvé (100%).
Échelle de conformité des articles et sous-articles avec 4
niveaux de conformité : informel [0-29%], aléatoire [30-59%],
convaincant [60-79%], conforme [80-100%].
Dès lors qu'un taux moyen de conformité de 60% est atteint pour
tous les articles d'une norme, une déclaration de conformité aux
exigences de cette norme est possible (déclaration établie selon
les exigences de la norme ISO 17050 [15]-[16].
L'utilisateur renseigne les métadonnées nécessaires et celles-ci
s'implémentent directement dans les autres onglets. Il peut
également modifier les échelles d'évaluation si nécessaire.
Figure 12 : onglet du mode d’emploi de l’outil (Source :
Auteurs)
{Résultats ISO 9001} / {Résultats ISO 13485} / {Résultats
ISO 14971} : Les résultats des niveaux de conformité
spécifiques aux exigences des normes sont représentés sous forme de
diagrammes radar et d'histogrammes. Ces graphes montrent en un coup
d’œil le niveau de conformité de chaque article et sous-article.
Cette présentation permet aux entreprises de visualiser facilement
les axes d'amélioration prioritaires.
{Résultats communs} : L'utilisateur y trouve les résultats
des 264 critères communs aux trois normes.
{Auto-déclaration de conformité ISO 9001}, {Auto-déclaration de
conformité ISO 13485}, {Auto-déclaration de conformité ISO 14971}
: ces trois onglets correspondent aux déclarations de
conformité du fournisseur faites selon les normes ISO 17050-1 et ISO
17050-2 [16]-[17].
Après avoir mesuré les niveaux de pratiques professionnelles au sein
de leur établissement grâce à l'outil "3-DIAG", les utilisateurs
peuvent éditer les "déclarations de conformité" correspondant à
chacune des trois normes.
Ces déclarations dites de 1ère partie ne correspondent pas à une
auto-certification aux normes mais elles donnent l'assurance que les
critères spécifiés pour les trois normes ont été évalués et sont une
première étape vers la certification. Les bonnes pratiques ISO 17050
recommandent que la personne validant les résultats de la
déclaration et que celle responsable de la déclaration soient
différentes et sans conflit d'intérêt.
Ces déclarations peuvent être utilisées par la Direction comme un
outil interne de communication, mettant en valeur l'implication,
l'adhésion et les efforts communs réalisés par les collaborateurs
pour atteindre le niveau de conformité attendu.
{Sources de valeurs}: cet onglet utilitaire contient les valeurs
permettant d'effectuer les calculs des taux de conformité.
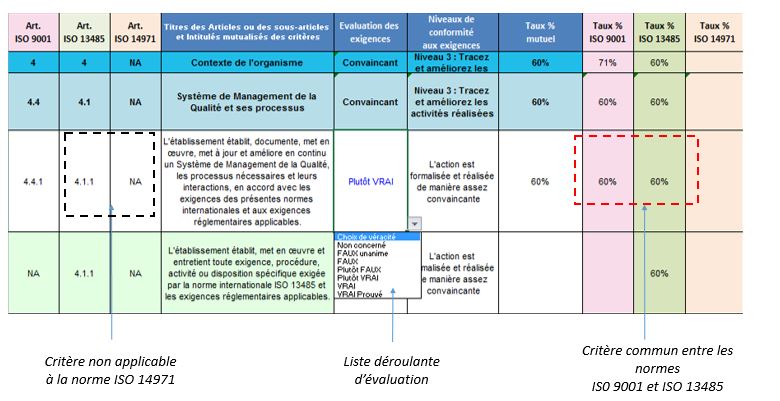
Les exigences reformulées sont retranscrites dans le tableau Excel
de l’onglet {Evaluation des exigences} et reliées aux chapitres et
paragraphes des normes concernées.
On retrouve ainsi seulement 491 critères au lieu de 745 si l’on
réalisait une évaluation individuelle selon chacune des normes.
Un code couleur est attribué en fonction du type d’exigence :
Type d’exigences
Couleur associée
Exigences communes aux normes
Bleu pâle
Exigences ISO 9001:2015
Rose
clair
Exigences ISO 13485:2016
Vert clair
Exigences ISO 14971:2013
Orange
Figure 14 : Tableau des
couleurs associées à chaque exigence(Source :
Auteurs)
Les articles et sous-articles de la norme ISO 13485:2016 sont mis en
exergue en bleu foncé.
L’utilisateur doit alors évaluer le niveau de réalisation de chaque
exigence. Pour cela, il utilisera l’échelle d’évaluation suivante :
Niveau de véracité
Taux (%)
Commentaire
Non-concerné
NA
L'action ne concerne pas l'établissement
évalué.
Faux unanime
0
L'action n'est pas réalisée selon l'avis de
tous les acteurs impliqués.
Faux
20
L’action est rarement réalisée ou de manière
aléatoire.
Plutôt faux
40
L'action est parfois réalisée mais de manière
informelle.
Plutôt vrai
60
L'action est formalisée et réalisée de
manière assez convaincante.
Vrai
80
L'action formalisée est toujours réalisée et
améliorée.
Une échelle de véracité à 6 niveaux a été privilégiée pour permettre
à l’utilisateur de nuancer sa réponse a fortiori car certaines
exigences ont été reformulées.
Chaque niveau de véracité des différentes exigences est ensuite
exploité. Une moyenne arithmétique des niveaux de conformité est
effectuée, de sorte à avoir une évaluation par grand chapitre.
Taux
Niveau de conformité
Commentaire
[0%-29%]
Insuffisant
Niveau 1 : Les activités doivent être
formalisées davantage.
[30%-59%]
Aléatoire
Niveau 2 : la bonne exécution des activités
doit être pérennisée.
[60%-79%]
Convaincant
Niveau 3 : les activités doivent être tracées
et améliorées.
[80%-100%]
Conforme
Niveau 4 : Niveau optimum-il faut continuer à
progresser et communiquer les résultats.
NA
Non Applicable
Non applicable: l'exigence n'est pas
applicable.
Figure 16: Tableau des niveaux de
conformité par chapitre [4]
III. Edition et
interprétation des résultats
1. Edition et analyse des
résultats
Une fois l’onglet 3 {Evaluation des exigences} dûment complété, il
est possible d’obtenir plusieurs feuilles de résultats :
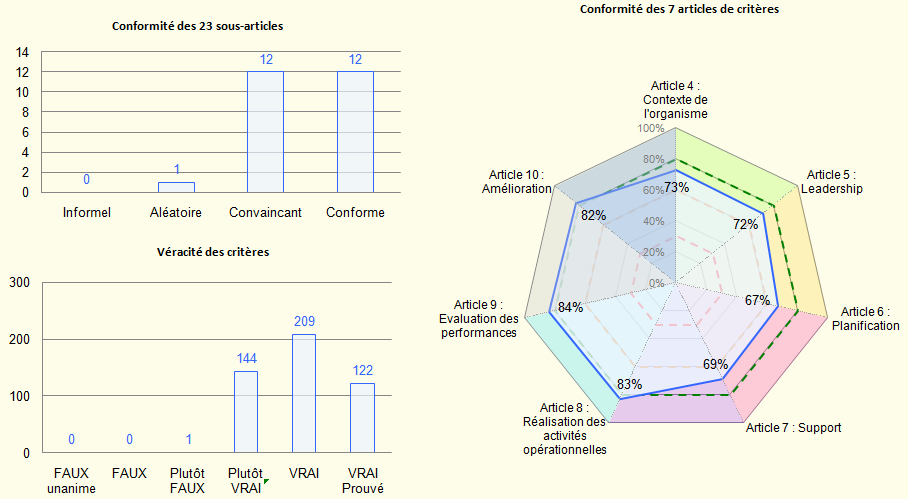
{Résultats communs} :
Le diagramme radar représente la conformité des articles communs aux
3 normes. Les valeurs des articles sont affichées à l’intérieur du
graphe, formant ainsi un polygone. Un tel type de diagramme donne
une vue d’ensemble générale du résultat. Il y a autant d’axes que
d'articles. Les articles (ou sous-articles) sont indiqués autour du
graphique. Les limites de conformité sont représentées par des
polygones en traits interrompus.
Les résultats de l’ensemble des sous-articles et des critères sont
représentés sous forme d’histogrammes. Le 1er histogramme représente
le niveau de conformité des sous-articles en indiquant le nombre des
sous-articles pour chaque niveau de conformité. Quant au 2ème, il
représente la véracité des critères d’exigence.
Ces diagrammes donnent une bonne vue d’ensemble de la situation.
Dans le même onglet, la présentation des résultats est détaillée
pour chaque sous article.
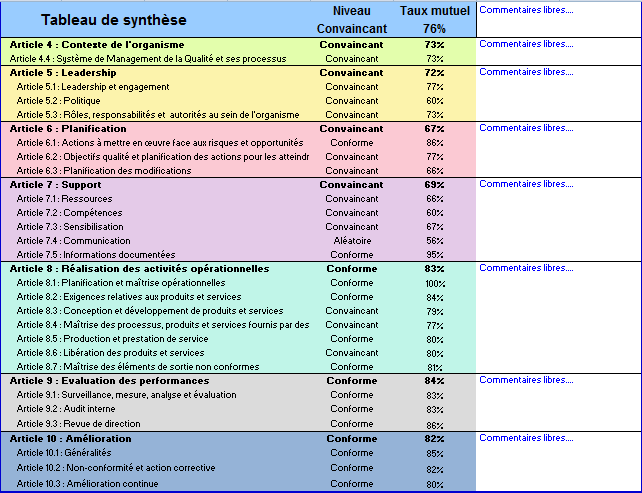
La synthèse des résultats des articles et des sous-articles est
présentée dans le même onglet sous la forme d’un tableau indiquant
le taux ainsi que le niveau de conformité.
Figure 18 : Tableau de synthèse
des résultats (Source : Auteurs)
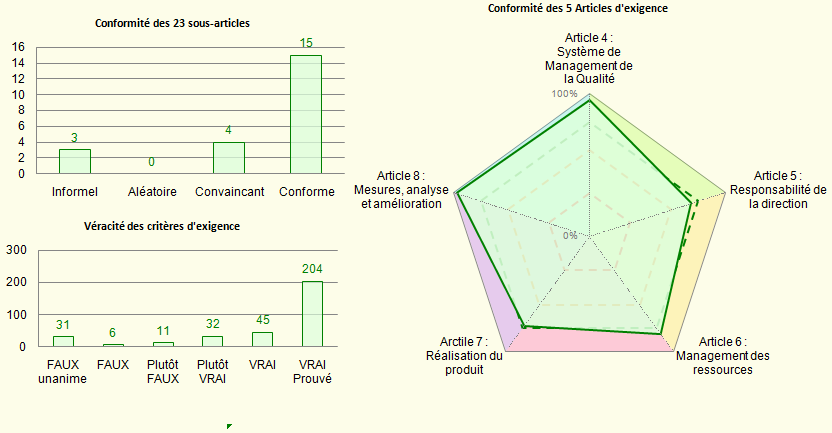
{Résultats ISO 9001} / {Résultats ISO 13485} / {Résultats ISO
14971} : Un onglet par norme pour décrire le niveau de conformité
spécifique à chaque norme.
Figure 19 : Présentation des
résultats de conformité pour chaque norme (Source
: Auteurs)
Pour aider l’utilisateur à bien respecter les exigences d’une telle
norme, des conseils ont été donnés à la fin de chaque onglet de
résultat. Ces conseils décrivent le contexte et la finalité de
chaque article ainsi que la personne responsable.
Figure 20 : Conseils pour
atteindre le niveau de conformité (Source
: Auteurs)
{Auto-déclaration de conformité ISO 9001}, {Auto-déclaration de
conformité ISO 13485}, {Auto-déclaration de conformité ISO 14971} :
Ces trois derniers onglets correspondent aux évaluations de
conformité – déclarations de conformité du fournisseur (NF EN
ISO/CEI 17050-1) pour chaque norme.
L’objectif de la déclaration est l’estimation de niveau de
conformité de chaque article aux exigences.
Un tel article est considéré "non-déclarable" s’il a un niveau
de conformité inférieur à 60%.
2. Synthèse sur les apports
de l’outil d’autodiagnostic
Figure 21 : logigramme de synthèse des apports de l’outil
d’auto-diagnostic (Source : Auteurs)
Le nouveau règlement européen des Dispositifs Médicaux qui entrera
en application en 2020 a renforcé les exigences essentielles
notamment en termes de gestion des risques, d’aspects cliniques et
de plan de surveillance du marché. Les entreprises vont devoir
s’adapter.
Elles devront pour cela s’appuyer sur la norme NF EN ISO 14155
Investigation clinique des Dispositifs Médicaux pour sujets humains
(Bonnes Pratiques Cliniques).
Par ailleurs, ce règlement précise les règles applicables aux
logiciels et renforce les exigences essentielles. Le développement
des logiciels et des applications mobiles dans le domaine de santé
est en pleine expansion. Ce développement peut varier d’un simple
logiciel de collecte et d’affichage des données à une application à
finalité complexe qui analyse, traite et affiche un certain nombre
de résultats. Pour être qualifié de DM ou de DM DIV, un logiciel
doit avoir une utilisation à des fins médicales [19].
Les fabricants doivent identifier la nature et la finalité médicale
des produits conçus de sorte à évaluer si le logiciel ou
l’application de santé relèvent du statut de DM ou de DM DIV. Pour
ce faire, ils doivent se baser sur la réglementation qui classifie
les applications et les logiciels selon leur destination d’usage :
• Un logiciel est considéré comme un dispositif
médical lorsqu’il permet de traiter des informations et des données
médicales aidant au diagnostic médical.
• Un dispositif de santé qui génère un résultat
suite à une simple recherche dans une base de données sans les
analyser ne répond pas à la définition du dispositif médical. Il est
considéré comme étant un logiciel de santé qui correspond à des
finalités médicales.
• Tout dispositif de télécommunication qui a pour
but d’échanger d’information médicale ou de réaliser des actes à
distance est considéré comme étant un logiciel de santé mais il ne
correspond pas à des finalités médicales, même s’il porte sur des
informations médicales.
Les entreprises pourront donc s’appuyer sur la norme NF EN 62304 :
Spécificité de la gestion du risque des logiciels DM ou DM DIV.
Les deux normes NF EN ISO 14155 Investigation clinique des
Dispositifs Médicaux pour sujets humains (Bonnes Pratiques
Cliniques) et NF EN 62304 : Spécificité de la gestion du risque des
logiciels DM ou DM DIV pourraient être transposées dans des outils
d'autodiagnostic, de sorte à pouvoir réaliser une évaluation du
système qualité par rapport à ces deux autres normes.
Conclusion
Les entreprises biomédicales doivent démontrer la conformité de
leurs dispositifs médicaux aux exigences essentielles et doivent
mettre en œuvre un système de management de la qualité et du risque.
Celui-ci est étayé par les normes ISO 9001, ISO 13485 et ISO 14971
qui lui confère une présomption de conformité aux exigences
essentielles telles qu’elles sont décrites par les Directives
européennes de la Nouvelle Approche. La conformité à ces trois
référentiels facilite l'obtention du marquage CE et améliore la
compétitivité de ces entreprises.
Ces travaux ont donc consisté à mutualiser les exigences des trois
normes et à concevoir l'outil "3-DIAG" pour permettre aux
entreprises de mesurer leurs niveaux de conformité aux trois normes
et de gagner du temps dans les processus de certification sur chacun
des référentiels.
L'outil "3-DIAG" permet une évaluation partielle ou totale de 491
critères (3h pour une évaluation totale au lieu de 8 heures environ
pour 745 exigences). Les résultats sont affichés sous forme de
tableaux, histogrammes et diagrammes radar afin d'en faciliter
l'interprétation visuelle et la compréhension rapide pour identifier
les plans d'actions prioritaires.
L'utilisation de cet outil permet d'améliorer la qualité des
produits et des services des entreprises biomédicales et d'assurer
la sécurité des utilisateurs des dispositifs médicaux et des
patients.
[1]AFNOR, « NF EN 62304-Logiciels de dispositifs
médicaux Processus du cycle de vie du logiciel ». EditionAfnor, www.afnor.org, oct-2006.
[2]« Règlement (UE) 2017/745 du Parlement européen et
du Conseil du 5 avril 2017 relatif aux dispositifs médicaux,
modifiant la directive 2001/83/CE, le règlement (CE) n°
178/2002 et le règlement (CE) n° 1223/2009 et abrogeant les
directives du Conseil 90/385/CEE et 93/42/CEE ». Journal
officiel de l’Union Européenne, http://eur-lex.europa.eu,
05-mai-2017.
[4]A. Kouiten, A. Harkani, H. B. Ben Charrada, S. Kambou,
N. Noulaquape, et T. J. Tchinde, « Nouvelle ISO 9001
(2015) et future ISO 13485 (2016) : Mutualisation des
exigences et outil bi-diagnostic pour la performance des
entreprises biomédicales », Université de Technologie de
Compiègne, Master Qualité et Performance dans les
Organisations (QPO), Master Technologies et Territoires de
Santé (TTS) et Mastère Spécialisé Normalisation, Qualité,
Certification, Essai (NQCE), Mémoire d’Intelligence
Méthodologique du projet d’intégration, https://www.utc.fr/master-qualite,
puis « Travaux » « Qualité-Management »
réf n°339, janv. 2016.
[5]AFNOR, « NF EN ISO 9001- Systèmes de management de
la qualité- Exigences ». EditionsAfnor, Paris, www.afnor.org, oct-2015.
[6]AFNOR, « NF EN ISO 13485- Dispositifs médicaux -
Systèmes de management de la qualité - Exigences à des fins
réglementaires ». EditionAfnor, www.afnor.org,
sept-2012.
[7]« NF EN ISO 14971 Dispositifs médicaux -
Application de la gestion des risques aux dispositifs
médicaux ». EditionsAfnor, Paris, www.afnor.org,
05-janv-2013.
[8]Syndicat National de l’Industrie et des Technologies
Médicales, « Panorama de la filière industrielle des
dispositifs médicaux en France en 2017 ». Edition Snitem,
Paris, www.snitem.fr, 2017.
[10]« RÈGLEMENT
(UE) 2017/ 746 DU PARLEMENT EUROPÉEN ET DU CONSEIL - du 5
avril 2017 - relatif aux dispositifs médicaux de diagnostic in
vitro et abrogeant la directive 98/ 79/ CE et la décision
2010/ 227/ UE de la Commission ». [En ligne]. Disponible
sur: http://eur-lex.europa.eu/legal-content/FR/TXT/PDF/?uri=CELEX:32017R0746&from=EN.
[Consulté le: 22-janv-2018].
[12]JPMA,
« Revision of Japanese Medical Device QMS requirements |
Pharmaceuticals and Medical Devices Agency », Japanese
Pharmaceuticals and Medical devices Agency. [En ligne].
Disponible sur: https://www.pmda.go.jp/english/review-services/regulatory-info/0004.html.
[Consulté le: 01-oct-2017].
[15]« NF
EN ISO/CEI 17050-1: Evaluation de la conformité - Déclaration
de conformité du fournisseur ». EditionsAfnor, Paris, www.afnor.org, 24-sept-2011.
[16]« NF
EN ISO/CEI 17050-2 - Évaluation de la conformité - Déclaration
de conformité du fournisseur - Partie 2 : documentation
d’appui ». EditionsAfnor, Paris, www.afnor.org,
01-avr-2005.
[17]El
Marsaoui, Lamkadem, Mancet, Meksi, « Référentiels qualité
majeurs pour les entreprises biomédicales », Université
de Technologie de Compiègne, Master Qualité et Performance
dans les Organisations (QPO), Master Technologies et
Territoires de Santé (TTS), Mémoire d’Intelligence
Méthodologique du projet d’intégration, https://www.utc.fr/master-qualite,
puis « Travaux » « Qualité-Management »
réf n°424, janv. 2018.




 sous
format .xls
sous
format .xls