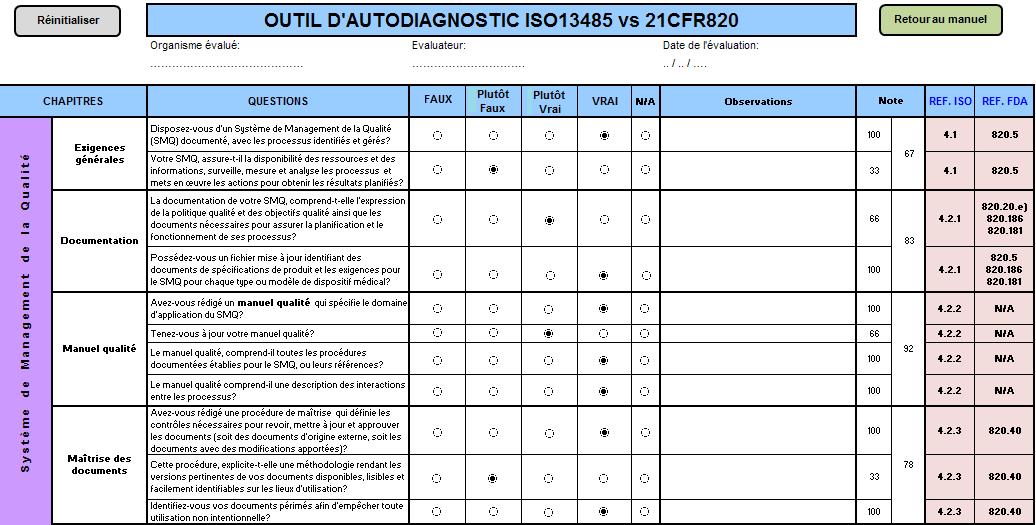
|
Avertissement
|
Si vous arrivez
directement sur cette page, sachez que ce travail est un rapport
d'étudiants et doit être pris comme tel. Il peut donc
comporter des imperfections ou des imprécisions que le lecteur
doit admettre et donc supporter. Il a été
réalisé pendant la période de formation et
constitue avant-tout un travail de compilation bibliographique,
d'initiation et d'analyse sur des thématiques associées
aux concepts, méthodes, outils et expériences sur les
démarches qualité dans les organisations. Nous ne faisons aucun usage commercial et la
duplication est libre. Si vous avez des raisons de contester ce droit
d'usage, merci de nous en faire part .
L'objectif de la présentation sur le Web est de
permettre l'accès à l'information et d'augmenter ainsi
les échanges professionnels. En cas d'usage du document,
n'oubliez pas de le citer comme source bibliographique. Bonne
lecture...
|
|
Autodiagnostic
ISO13485 et
21CFR820
(Système de Management de la
Qualité pour les Dispositifs Médicaux)
|

Anas RAIS
|

Samantha
GLASGOW |
|

Nicole
TAVARES
DE MELO
|
Référence bibliographique
à rappeler pour tout usage :
Autodiagnostic
ISO13485 et 21CFR820
(Système de Management de la Qualité pour les Dispositifs
Médicaux)
Samantha GLASGOW, Anas RAIS,
Nicole TAVARES DE MELO, Adrian PARTEARROYO
Responsable: Gilbert
FARGES
Projet
d'Intégration
MASTER Management de la Qualité (MQ), UTC,
2009-2010 URL : https://www.utc.fr/master-qualite
; Université de
Technologie de Compiègne
|
|
RÉSUMÉ
Pour aider les entreprises impliquées dans la vente des
dispositifs médicaux dans le marché Européen ainsi
qu’exporter aux Etats-Unis, un outil d’autodiagnostic a
été développé à partir de la
réglementation 21CFR820 (Medical Devices – Quality System
Regulation) de la Food and Drug Administration (FDA) et la norme
ISO13485:2004 (Dispositifs médicaux - Systèmes de
Management de la qualité).
Pour une entreprise européenne, un dispositif médical
doit faire preuve de performance tout en assurant la
sécurité des patients et des utilisateurs, cela passe par
l’obtention du marquage CE. Ce marquage impose à l’entreprise
d’avoir un système de management de la qualité qui peut
être obtenu en répondant aux exigences de la norme ISO
13485. Cependant, si une entreprise souhaite exporter ses dispositifs
médicaux aux Etats-Unis, la norme ISO 13485 n’est plus
suffisante et l’organisme doit répondre aux exigences
supplémentaires de la FDA par la réglementation 21CFR820.
Mots clés : DM,
Dispositifs Médicales, Système de management, ISO 13485,
21CFR820, Marquage CE, Exigences, FDA.
Télécharger l'outil
d'autodiagnostic ici : autodiagnostic_iso13485_21cfr820_v1.6.xls
|
|
ABSTRACT
In order to help the companies involved in the production and
the sale of medical devices in the European market and also to export
to the United States of America, an autodiagnostic tool has been
developed comparing the FDA’s (Food and Drug Administration) 21CFR820
regulation (Medical Devices – Quality System Regulation) with the
ISO13485:2004 (Medial Devices : Quality Management Systems) standard.
For an European company, a medical device has to do a performance test
to ensure the patient’s and user’s security. This device must obtain
the CE labeling. This labeling process imposes that the company has a
quality management system that can be obtained by responding to the
requirements of the ISO13485:2004 standard. However, if a company wants
to export its medical devices to the USA, then the ISO13485:2004
standard is not enough and the organization has to satisfy the
additional requirements of the FDA’s 21CFR820 regulation.
Key words : DM, Medical
Devices, Management system, ISO 13485, 21CFR820, CE labeling,
Requirements, FDA.
Download the autodiagnostic tool
here : autodiagnostic_iso13485_21cfr820_v1.6.xls
|
|
RESUMEN
Para poder ayudar a las empresas implicadas en la
producción y venta de dispositivos médicos en el mercado
Europeo y tengan intenciones de hacer exportaciones a los Estados
Unidos de Norte América, se ha desarrollado una herramienta de
Auto Diagnostico a partir de la comparación entre la
reglamentación 21CFR820 (Medical Devices – Quality System
Regulation) de la Food and Drug Administration (FDA) y de la norma
ISO13485:2004 (Medial Devices : Quality Management Systems).
Para una empresa Europea, un dispositivo médico debe pasar una
prueba de eficiencia, todo asegurando la seguridad de los pacientes y
de los usuarios del dispositivo, este pasa por la obtención del
marcado CE. Este marcado impone a la empresa un sistema de manejo de
calidad que puede ser obtenido respondiendo a las exigencias de la
norma ISO 13485. Sin embargo, si una empresa desea exportar sus
dispositivos médicos a EU, la norma ISO 13485 ya no es
suficiente y el organismo debe responder a las exigencias
suplementarias de la FDA por la reglamentación 21CFR820.
Palabras Clave: DM,
Dispositivos Médicos, Sistema de manejo, ISO 13485, 21CFR820,
Marcado CE, Exigencias, FDA.
Descargar la herramienta de autodiagnostico aquí : autodiagnostic_iso13485_21cfr820_v1.6.xls
|
REMERCIEMENTS
La réalisation de ce travail n'aurait pas été
possible sans le concours de certaines personnes que nous tenons
à remercier :
Monsieur Gilbert FARGES et Monsieur Jean Pierre CALISTE, responsables
respectivement des formations « Management de la
qualité» et « NQCE », pour leurs conseils
avisés et le suivi de notre projet.
Madame Laetitia BARDY, responsable qualité à Hutchinson
Santé, pour ses conseils et son aide lors de la
réalisation de notre outil.
Monsieur Thiago Barbosa, ancien étudiant Master UTC, pour son
aide à la compréhension des démarches de la norme
ISO et de la réglementation FDA.
Les entreprises ayant répondu à notre questionnaire
lors de notre enquête « impact outil ».
SOMMAIRE
RÉSUMÉ
ABSTRACT
RESUMEN
Remerciements
Sommaire - Table des illustrations
GLOSSAIRE
ISO : International Standard
Organisation (Organisation Internationale de Normalisation)
DM : Dispositifs Médicaux
CFR : Code of Federal Regulations
FDA : U. S. DEPARTEMENT OF HEALTH AND HUMAN SERVICES – FOOD AND DRUG
ADMINISTRATION
AMDEC : Analyse des Modes de Défaillance, de leurs Effets et de
leur Criticité
PDS : Planification Dynamique Stratégique
SNITEM : Syndicat National de l'Industrie des TEchnologies
Médicales
1.
Contexte,
enjeux
et
problématique:
Dans le cadre de l’Unité
d’Enseignement QP10, nous devons réaliser un projet
d’intégration. Ce projet porte sur les dispositifs
médicaux et plus particulièrement sur le Système
de Management de la Qualité. Au niveau européen, il
s’agit de la norme ISO 13485 (Dispositifs médicaux -
Système de Management de la qualité). Cependant, si une
entreprise souhaite exporter ses dispositifs médicaux aux
États-Unis, la norme ISO 13485 ne suffit plus. Il faut donc que
l’organisme réponde aux exigences supplémentaires de la
FDA par la réglementation 21CFR820 (Médical Devices –
Quality
System Regulation).
Afin de comprendre ce qu’ est un
dispositif médical, nous avons repris la définition de la
norme ISO 13485, selon laquelle un Dispositif médical est:
« tout instrument, appareil, équipement, machine,
dispositif, implant, réactif in vitro ou calibreur, logiciel,
matériel ou autre article similaire ou associé, dont le
fabricant prévoit qu’il soit utilisé seul ou en
association chez l’être humain […] et dont l’action principale
voulue dans ou sur le corps humain n’est pas obtenue par des moyens
pharmacologiques ou immunologiques ni par métabolisme, mais dont
la fonction peut être assistée par de tels moyens ».
Il est intéressant de noter que
l’ISO 13485 :2003 a été fondée sur l’ISO 9001
:2000. C’est pourquoi, nous nous aiderons de l’ancien projet UTC
sur la norme ISO 9001 réalisé en 2008.
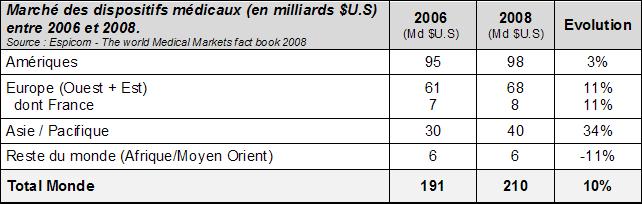
Le marché mondial des
Dispositifs Médicaux est très important puisqu’en 2008,
l'industrie mondiale des dispositifs médicaux est estimée
à 210,2 milliards de U.S $ (166,7 milliards d'€). Ce
marché est en constante augmentation puisqu’il a connu une
progression d'environ 10% entre 2006 et 2008.
Tableau
1 : Marché des
Dispositifs Médicaux en 2006 et 2008 au niveau mondial
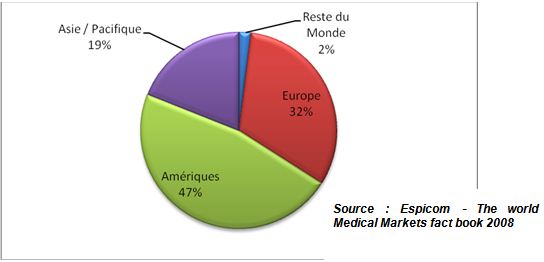
Au niveau des marchés mondiaux, le marché européen
des dispositifs médicaux est le deuxième plus grand
marché au monde, derrière le marché
américain et devant le marché japonais. Il
représente 30% des ventes mondiales, pour une valeur
estimée à 54,8 milliards d'euros. Extrêmement
concurrentielle, l'industrie des dispositifs médicaux repose sur
une recherche constante d'innovation ; elle emploie près de 385
000 personnes (Source : Eucomed Industry Profile 2003).
Figure
1 : Marchés mondiaux des
dispositifs médicaux en 2008
1.3.
Problématique
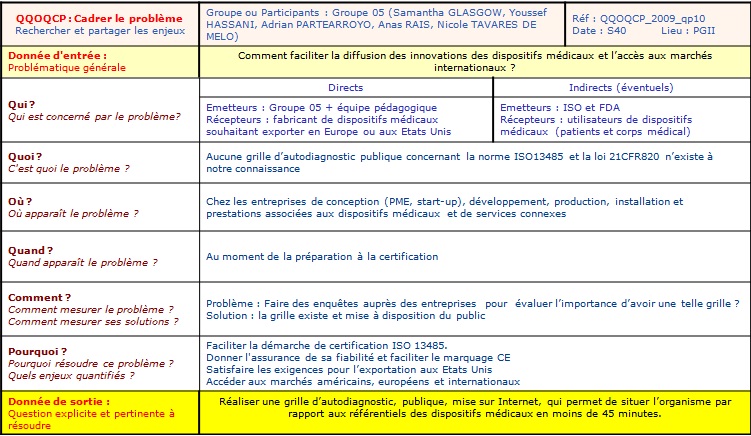
A ce jour, la démarche d’obtention d’une certification pour un
organisme est longue et fastidieuse. Un outil d’autodiagnostic
s’avère indispensable pour faciliter cette démarche. A
notre connaissance, aucun outil d’autodiagnostic public combinant la
norme ISO 13485 et la réglementation 21CFR80 n’existe. Afin de
mieux cerner notre problématique, nous avons
réalisé un QQOQCP :
Tableau
2 : Grille de synthèse du
QQOQCP
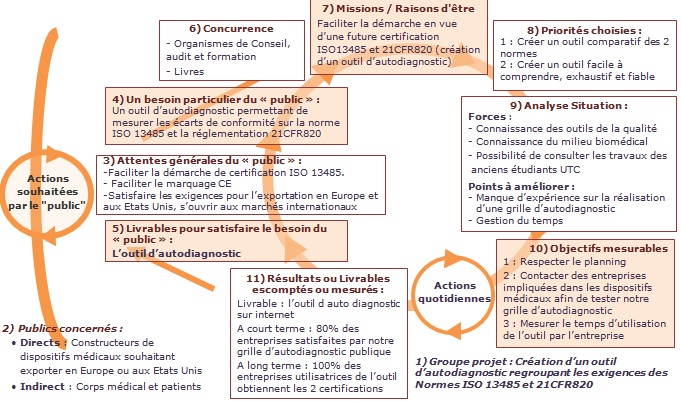
Notre outil intéressera
principalement aux entreprises qui conçoivent et vendent des
dispositifs médicaux. Afin de mieux visualiser ces objectifs,
nous avons procédé à un Plan Dynamique
Stratégique :
Figure
2 : Planification Dynamique
Stratégique
Nos priorités concernant l’outil seront la facilité
d’appropriation (intuitif), la facilité de compréhension
(questions claires), le temps d’utilisation (1 à 2h maximum) et
la fiabilité (bon respect de la norme ISO 13485 et de la
réglementation 21CFR820).
1.4.
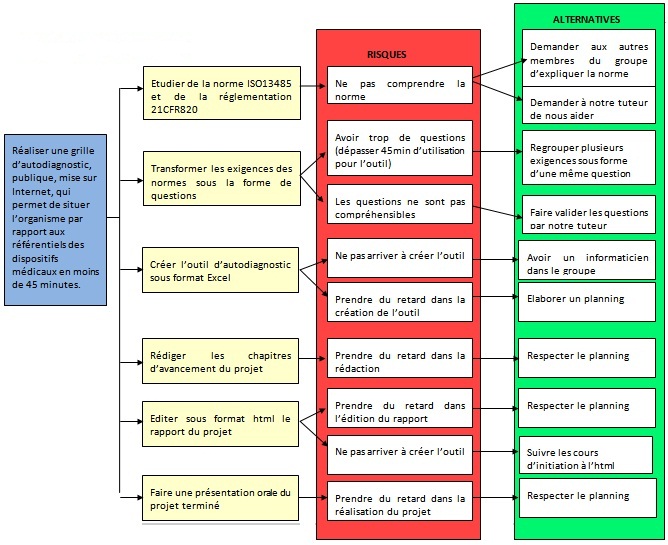
Risques projet
Pour identifier les risques du projet, nous avons réalisé
un diagramme de décisions. Cela consiste à partir de
notre problématique et de déterminer nos plans d’actions,
puis d’identifier pour nos actions, les risques associés. Une
fois les risques identifiés, nous cherchons des alternatives
permettant de palier au risque.
Diagramme
1
: Arbre des
décisions
2. Enquête
concernant l’impact de l’outil
2.1. Objectifs
de l’enquête:
Pour avoir des notions concrètes
sur le contexte d’application de notre projet, une enquête a
été crée. L’objectif de cette étape est de
mieux connaître l’impact de l’outil et les besoins des clients,
ainsi qu’à partir des réponses obtenues, trouver un
contexte qui soit favorable à l’utilisation de l’outil
concerné par ce projet.
2.2. Réalisation
de l’enquête:
Le questionnaire crée comprend
quatre questions à choix multiples, à répondre
par OUI ou par NON, concernant les quatre points suivants :
• Conformité
de l’entreprise aux normes;
• Type de service utilisé pour la mise en
œuvre d’un système de management de la qualité dans
l’entreprise;
• Exportation vers le marché
nord-américain;
• Préférences sur la forme de l’outil.
A partir du site du SNITEM, et des contacts des anciens
étudiants du Master Management de la Qualité, 144
entreprises ayant des activités liées aux dispositifs
médicaux ont été sélectionnées et
contactées par courrier électronique. Nous avons
reçu treize réponses (figure3). Les entreprises ayant
répondu sont les suivantes :
EUROS
|
FRANCEMED
|
HUTCHINSON SANTE
|
INOMED
|
MAQUET
|
RESMED
|
AXES VISION TECHNOLOGY
|
PHILIPS SYSTEMES MEDICAUX
|
STRYKER ORTHOPAEDICS CAEN
|
LIFE PARTNERS EUROPE
|
OTTO BOCK FRANCE
|
PETERS SURGICAL
|
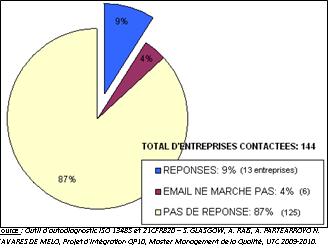 Figure 3 :
Taux de réponses pour
l’enquête
Figure 3 :
Taux de réponses pour
l’enquête
retour sommaire
2.3. Résultats
de l’enquête:
Les résultats obtenus permettent
d’élaborer une représentation graphique de l’état
des entreprises par rapport au contexte et de tirer quelques
conclusions sur leurs situations et leurs préférences
pour l’outil. A partir de cette analyse nous trouvons que cette grille
d’autodiagnostic peut être utile chez les industriels
biomédicaux :
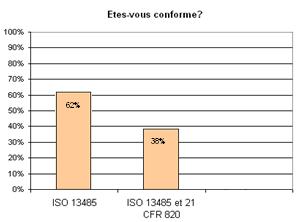
a. La plus part des entreprises (62%) sont déjà
certifiés ISO 13485, par contre, ne sont pas conformes aux
exigences de la réglementation 21CFR820 de la FDA (figure 4).
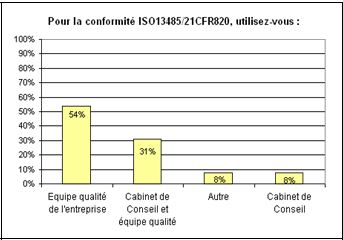
b. 54% des entreprises utilisent les services d’une équipe
qualité interne afin de se préparer pour répondre
aux exigences de l’ISO et de la FDA. Ceux sont ces équipes qui
utiliseront notre outil. (figure 5)
Figure
4 : Question n° 1 de
l’enquête. (source [16]) Figure 5 : Question n°2 de
l’enquête (source [16])
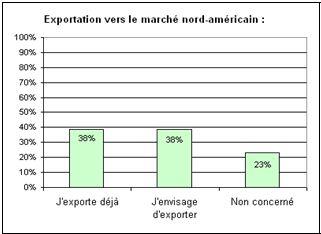
c. 38% des
industriels envisagent d’exporter vers le marché
nord-américain et ont donc besoin d’être conforme aux
exigences de la FDA (figure 6).
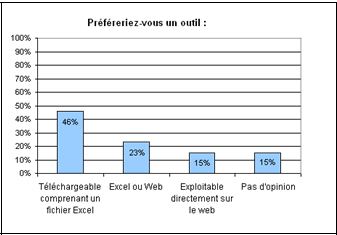
d. Concernant la forme de l’outil, la plupart de ceux
qui ont répondu à l’enquête ont une
préférence pour un outil téléchargeable
comprenant un fichier Excel (figure 7). Cette information est
importante pour la suite de ce projet.
Figure
6 : Question n°3 de
l’enquête (source [16]) Figure 7 : Question n°4 de
l’enquête (source [16])
3.
Elaboration des options d’intervention pour l’outil
Concernant la conception de l’outil
d’autodiagnostic, trois options ont été pensées et
analysées par rapport aux avantages, inconvénients,
faisabilité, pertinence, risques et alternatives
déterminées. A partir de cette étude, nous avons
pu choisir l’option d’intervention la plus pertinente à mettre
en place.
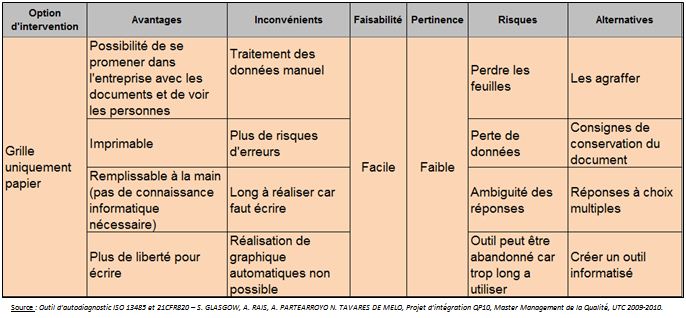
Cette option est certes facile à
faire en manière de conception puisqu’il s’agit juste d’utiliser
un logiciel de traitement de texte mais cette option est peu pertinente
pour les entreprises et pour nous car il existe
déjà sur le marché des grilles d’autodiagnostic
papier.
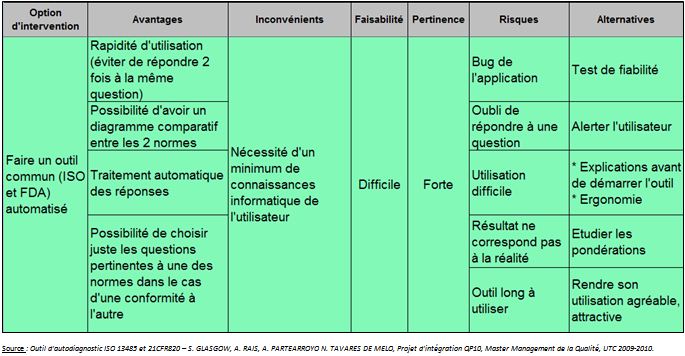
Option 2: Deux outils
automatisés
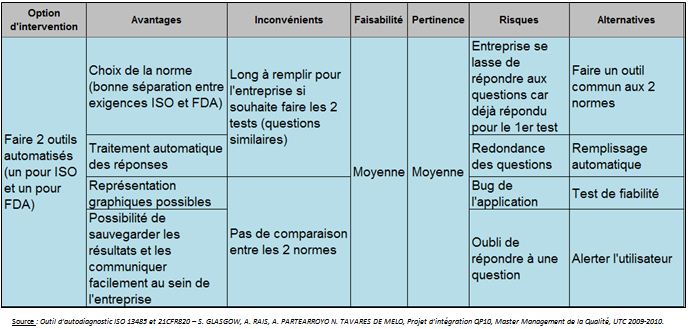
Nous avons considéré cette option comme ayant une
pertinence moyenne car si l’utilisateur souhaite tester les 2 outils,
cela sera long pour lui et ennuyeux dû à la
répétition des questions communes pour la norme ISO
et la réglementation 21CFR820. Néanmoins, l’avantage de
cet outil automatisé est qu’il permet de réaliser des
graphiques à partir des résultats obtenus et donc de
permettre à l’utilisateur de visualiser en un coup d’œil
où l’entreprise possède des lacunes.
Option 3: Un outil
automatisé commun à l’ISO13485 et la 21CFR820
Cette option a été retenue car elle permet de combiner
à la fois la norme ISO et la réglementation 21CFR820 dans
un même outil. L’utilisateur pourra lui-même choisir s’il
souhaite répondre aux questions concernant la norme ISO ou la
réglementation 21CFR820 ou bien les deux.
Après avoir
choisi l’option 3
(outil automatisé commun à la norme ISO et 21CFR820),
nous nous sommes posé la question : « comment faire pour
mettre en œuvre cette option » ? Nous avons pensé à
trois façons de procéder:
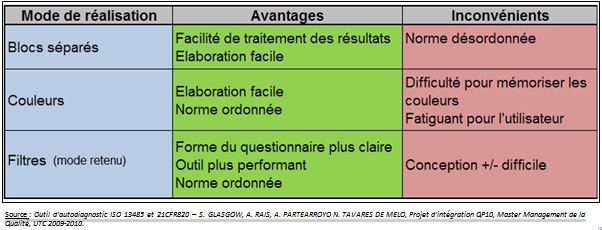
Le mode de réalisation «
blocs séparés » correspond à un bloc qui
comprendrait la norme ISO, un bloc la réglementation FDA et un
bloc qui regrouperait les questions communes aux deux. Mais
l’inconvénient est que la norme serait désordonnée
puisque les chapitres de l’ISO ne sont numérotés de
la même manière que la 21CFR820.
Le mode de réalisation «
couleurs » correspond au mélange des exigences de la norme
ISO et 21CFR820. Les exigences seraient regroupées et donc
ordonnées mais afin de repérer la norme ISO et la
21CFR820, nous metterions des couleurs pour chaque norme. Cette
méthode serait fatiguante visuellement pour l’utilisateur.
Le dernier mode de réalisation
correspond à l’utilisation de filtres dans Excel. L’utilisateur
n’aura qu’à choisir en cliquant sur un bouton s’il veut
répondre aux exigences de la norme ISO ou de la
réglementation 21CFR820. C’est selon ce mode que nous allons
réaliser notre outil d’autodiagnostic.
4.
Réalisation de
l’outil d’autodiagnostic
4.1. Les
exigences de la norme ISO13485 et de la réglementation 21CFR820
Pour réaliser les questions de l’outil, nous avons lu plusieurs
fois la norme ISO 13485 et la réglementation américaine
21CFR820 afin de bien comprendre les exigences de celles-ci.
A partir de là, nous avons regroupé les exigences
communes à l’ISO13485 et à la 21CFR820 car notre objectif
était d’avoir un seul et même outil pour ces deux normes.
Cela permet de diminuer le nombre de questions à répondre
pour l’utilisateur et d’éviter la redondance.
Après avoir réussi à regrouper les exigences
communes, nous les avons transformées en questions.
Une fois les questions faites, nous les avons listées en suivant
l’ordre des chapitres de l’ISO13485. Suite à cela, nous avons
ajoutés les quelques questions propres à la
réglementation américaine 21CFR820 dont le chapitre
correspondait à la norme ISO13485. Cela nous a mené
à 121 questions, or, au départ il était
prévu que notre outil soit rapide d’utilisation (45 minutes
maximum). Nous avons donc demandé conseil auprès d’un
professionnel qui avait été sollicité lors de
notre enquête « impact outil » et qui avait
été d’accord pour tester notre grille, afin de savoir
s’il était préférable de privilégier la
rapidité de l’outil ou l’exhaustivité des exigences. Sa
réponse à été favorable concernant
l’exhaustivité. Suite à cela, nous nous sommes
lancés dans la réalisation Excel de l’outil.
4.2.
Réalisation
de
l’outil
sous
format
Excel
Lors du chapitre 2, nous avions énuméré plusieurs
possibilités pour réaliser notre outil
d’autoévaluation (cf.3.1. Mode de réalisation), l’option
retenue avait été l’utilisation de filtres afin de
permettre à l’utilisateur de choisir s’il souhaité
répondre aux exigences de la 21CFR820, à celles de
l’ISO13485 ou bien aux deux.
4.2.1.
L’Outil :
Pour réaliser l’outil, nous nous sommes inspirés de celui
réalisé en 2007 (Projet n°58 « démarche
ISO 9001 pour Master Qualité ») par des anciens de l’UTC.
L’outil final s’agit d’une grille de 121 questions comprenant les
critères les plus importants des deux documents
étudiés. Les questions sont ordonnées par rapport
aux chapitres de la norme ISO, en ajoutant les points
spécifiques de la réglementation de la FDA. L’outil a
été conçu de manière que l’utilisateur
puisse cliquer sur un bouton et choisir parmi 4 tests différents
à réaliser selon leurs besoins et profil de l’entreprise :
- Evaluation de conformité ISO et FDA : Ce test
permet au client d’évaluer sa conformité selon les
exigences de la norme ISO 13485 et de la réglementation de la
FDA 21CFR820. Dans ce cas, toutes les questions de la grille doivent
être répondues. Cette option s’applique aux entreprises
qui ne possèdent pas la certification ISO pour les DM, ou celles
qui veulent faire une nouvelle évaluation de leur système
de management de la qualité.
- Evaluation de conformité ISO : Le client
répond aux questions concernant uniquement la norme ISO
13485 ainsi que les questions communes entre cette norme et la
réglementation de la FDA. Cela leur permet d’évaluer leur
niveau de conformité à la norme ISO, cependant ne
s’applique pas aux entreprises qui veulent répondre aux
critères spécifiques de la FDA.
- Evaluation de conformité FDA : Ce test
contient toutes les questions communes à la norme ISO et
à la réglementation de la FDA ainsi que les questions
exclusives de la réglementation américaine. Sont
retirées les questions exclusives de l’ISO. Il est applicable
aux entreprises qui peuvent être déjà
certifiées ISO, mais qui veulent s’évaluer concernant la
réglementation de la FDA complète.
- Evaluation de conformité à la FDA
(exclusivement) : Est applicable uniquement aux entreprises qui sont
déjà certifiés ISO 13485. Il s’agit d’une
évaluation rapide, qui contient uniquement les questions
spécifiques de la réglementation 21CFR820 et qui n’ont
pas de correspondance avec les exigences de la norme ISO. Ce test part
du principe que les entreprises déjà certifiés ISO
sont conformes aux exigences plus importantes de cette norme. Cette
conclusion nous permet de réaliser un diagnostic rapide
concernant les exigences de la FDA.
4.2.2. L’auto diagnostic :
L’auto diagnostic est réalisée après le
traitement des réponses fournies par le client. La grille se
présente sur la forme d’un fichier Excel, où les macros
doivent être autorisées et où, pour chaque
question, l’outil présente une case
« observations », pour les commentaires
écrits et cinq boutons cliquables pour la réponse qui
concerne la situation de l’entreprise par rapport aux questions
posées. Chaque réponse cochée compte une certaine
quantité de points à l’évaluation finale :
- Faux – 0 %
- Plutôt faux – 33 %
- Plutôt vrai – 66 %
- Vrai – 100 %
- N/A – ne compte pas de points, élimine ce critère
des calculs.
A tout moment, l’utilisateur peut connaître les
références ISO et FDA associées à la
question.
Une fois que l’utilisateur a fini de répondre aux
critères, le questionnaire doit être validé. Dans
cette étape, l’outil vérifie si toutes les questions ont
été répondues. Dans le cas positif, le test est
complet et les diagrammes du résultat de
l’auto-évaluation sont présentés au client. Dans
le cas contraire, un message d’erreur s’affiche en demandant au client
de finir le remplissage du questionnaire.
4.3.
Bilan
sur
l’outil :
Téléchargement
direct
L’outil final contient 121 questions et demande environ une heure et
demie pour être répondu. Ces questions reprennent les
exigences les plus importantes des deux documents travaillés.
La présentation graphique comparative des résultats ainsi
que le caractère imprimable de l’outil ont été
réalisés comme initialement prévu pour l’outil
final.
Source
[16]
Figure 8 : Outil
d’autodiagnostic pour la qualité en conception des dispositifs
médicaux Norme ISO 13485 et réglementation FDA 21CFR820.
Une fois que l’utilisateur a fini de répondre aux questions,
la grille peut être validée. Dans cette étape,
l’outil vérifie qu’il a été répondu
à toutes les questions. Dans le cas positif, le test est complet
et la feuille de synthèse des résultats est
présentée et imprimable au format A4 (figure 9). Dans le
cas contraire, un message d’erreur s’affiche en demandant de finir le
remplissage du questionnaire.
La feuille de synthèse des résultats présente un
graphique radar ainsi qu’un tableau avec les notes en pourcentage de
validation obtenues pour chaque chapitre de l’ISO 13485 et de la
réglementation 21CFR820. Un code couleur fait rapidement
percevoir les points critiques.
L’importance relative des résultats pour chaque chapitre de
l’ISO peut être paramétrée par l’utilisateur (en
fonction de son contexte ou de ses priorités industrielles) en
modifiant les coefficients mis par défaut (figure 9).
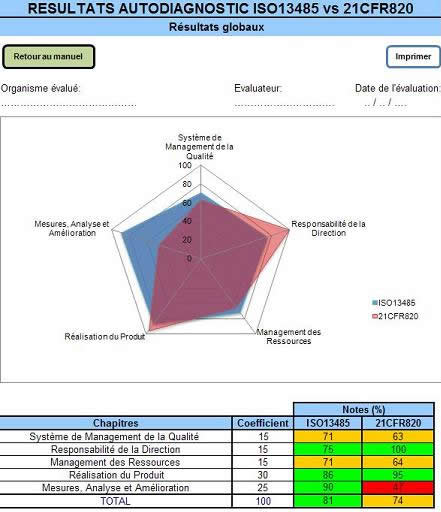
Source
[16]
Figure
9 : Vue d’un exemple des
résultats graphique de l’outil.
5.
Proposition d’amélioration de la grille suite aux tests en
entreprises
Une fois l’outil prêt, nous avons contacté quelques
entreprises afin de tester le produit final mais dû à
l’emploi du temps des responsables qualité, il ne nous a pas
été possible de le faire avant le Jalon 3.
En ce qui concerne la pertinence des questions, avant la
réalisation de l’outil nous les avons soumises à
l’opinion d’un professionnel de la qualité dans le domaine
biomédical. Quelques remarques on été faites et
nous les avons prises en compte.
La première démarche à effectuer sera donc de
tester l’outil chez un client, et d’accompagner l’utilisation au
coté du responsable en charge de le répondre. Une fois
que l’outil aura été testé, l’utilisateur pourra
répondre à un
questionnaire de satisfaction, afin de mesurer sa
satisfaction
face à l’outil selon différents aspects. Les
réponses seront recueillies dans une base de données qui
affichera l’opinion globale de la satisfaction de nos clients. A partir
des ces résultats il sera possible de prévoir d’autres
améliorations à l’outil actuel.
Conclusion
Le marché des dispositifs médicaux étant de
plus en plus concurrentiel et en plein développement, il est
important que les entreprises soient conformes à la norme ISO
13485 et à la réglementation FDA 21CFR820 afin de pouvoir
accéder, et éventuellement s’imposer, sur les
marchés Européen et Américain, améliorer
leurs produits et services et assurer la sécurité et le
respect des réglementations.
La démarche pour l’entreprise afin d’être conforme
à la norme ISO 13485 et à la réglementation
FDA 21CFR820 passe par un bilan de performance de son système de
management de la qualité.
L’outil proposé, basé sur les besoins des entreprises
estimés à partir d’une enquête
réalisée en Octobre 2009, leur permet de s’évaluer
rapidement (1 à 2 heures) par rapport à la norme ISO
13485 et à la réglementation FDA 21CFR820 en
répondant aux questions d’une grille d’autodiagnostic. Des
résultats graphiques montrent les forces et faiblesses de
l’entreprise et lui permet de mettre en application un plan d’action en
vue d’améliorer sa situation.
Avoir un système de management de la qualité selon les
exigences des normes et réglementations en vigueur permettra non
seulement d’accéder à des marchés importants pour
développer l’entreprise, mais aussi de garantir la
qualité et la sécurité des dispositifs
médicaux au bénéfice de celles des soins
délivrés au patient.
Références
Bibliographiques
Sites Internet :
[1] SGS, organisation d’inspection, contrôle et analyse de la
certification :
http://www.fr.sgs.com/fr/medical_devices?serviceId=28156&lobId=19719
, consulté le 8/10/09
[2] Ordre de pharmacien :
http://www.ordre.pharmacien.fr/fr/bleu/index4_3.htm,
consulté
le 30/09/09
[3] Quali-Conseil, Cabinet de conseil, audit et formation :
http://www.quali-conseil.com/iso_13485.html,
consulté
le 01/09/09
[4] Axess, Cabinet de conseil, audit et formation :
http://www.axess-qualite.fr/iso-13485.html,
consulté
le 01/09/09
[5] Qualitycoach, boutique spécialisée dans les produits
pour la Qualité :
http://www.qualitycoach.net/shop/shopexd.asp?id=5860,
consulté
le 06/09/09
[6] SNITEM, Syndicat national de l'industrie des technologies
médicales :
http://www.snitem.fr,
consulté le 12/10/09
Documents officiels :
[7] Dispositifs médicaux - Systèmes de management de la
qualité - Exigences à des fins réglementaires. NF
EN ISO 13485. Paris : AFNOR, 2004, 75 p.
[8] Quality System Regulations for Medical Devices. 21CFR820.1, CODE OF
FEDERAL REGULATIONS, U. S. DEPARTEMENT OF HEALTH AND HUMAN SERVICES –
FOOD AND DRUG ADMINISTRATION.
Révisé le 1er Avril 2001, Chapitre 1, Part 820, Titre 21,
Vol 8.
[9] Directive 93/42/CEE du Conseil, du 14 juin 1993, relative aux
dispositifs médicaux. LE CONSEIL DES COMMUNAUTÉS
EUROPÉENNES. Journal officiel n° L 169 du 12/07/1993 p. 0001
– 0043.
Thèse :
[10] Le dispositif médical : aspects réglementaires et
économiques. Evolution sur les dix dernières
années. POYET A. Thèse de Doctorat en Pharmacie. Lyon :
UNIVERSITE CLAUDE BERNARD – LYON I, 2003, 106 p.
Anciens travaux UTC :
[11] Intégration des exigences du 21 CFR 820 dans le processus
maintenance. DORMARD Laurent. Rapport de stage du Master management de
la qualité. Compiègne : Université de Technologie
de Compiègne, 2009, 43 p.
[12] Comment intégrer un nouveau produit dans un système
qualité ? BOULANGER Julien. Rapport de stage du Master
management de la qualité. Compiègne : Université
de Technologie de Compiègne, 2009, 55 p.
[13] Application de la norme 60601-1-6 pour évaluer l’aptitude
à l’utilisation des éclairages opératoires. &
Optimisation du système qualité de MAQUET SA. BARBOSA
Thiago. Rapport de stage du Master management de la qualité.
Compiègne : Université de Technologie de
Compiègne, 2009, 55 p.
[14] La mise en place de la certification ISO 13485 au sein d’AXESS
Vision Technology. HERMELIN Anne. Rapport de stage du Master management
de la qualité. Compiègne : Université de
Technologie de Compiègne, 2009, 46 p.
[15] Réaliser une grille d’audit ISO 9001 : 2000 simple
d’utilisation et associant des outils graphiques.BOURGET J., HAMDOUCH
H., MORET R. Projet d’intégration QP10 du Master management de
la qualité. Compiègne : Université de Technologie
de Compiègne, 2009, 7 p.
[16] AutodiagnosticISO13485 et 21CFR820 (Système de Management
de la Qualité pour les dispositifs médicaux) – Glasgow
S., Rais A., Partearroyo A., Tavares de Melo N., Projet
d’intégration Master Management de la Qualité, 2009-2010.
Université de Technologie de Compiègne, https://www.utc.fr/master-qualite/,
rubrique
« Travaux », n°124.
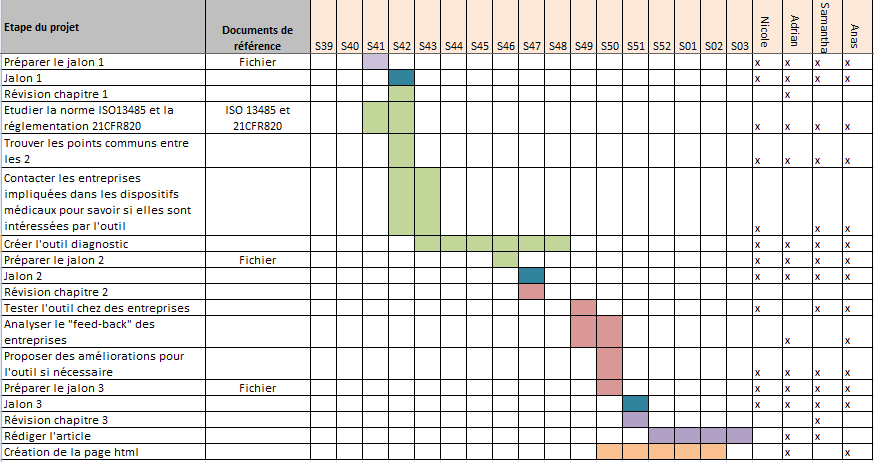
Annexe I : Planning du projet
Annexe
II : e-mail
envoyé aux entreprises pour l’enquête « impact outil
»
Bonjour,
Pour permettre à ... (nom de l’entreprise) … de se situer par
rapport à la norme
ISO 13485 et la réglementation 21CFR820, une équipe
projet de l’UTC
réalise un outil d’auto-évaluation qui sera mis
gratuitement à votre
disposition sur internet.
Pour mieux situer l’impact de l’outil et la satisfaction de vos
Besoins, merci de répondre aux questions ci-dessous :
1) Etes-vous certifiés : (OUI / NON / Ne connait pas)
-ISO13485
-21CFR820
2) Pour obtenir la certification ISO13485/21CFR820, utilisez-vous
(OUI/NON) :
-Cabinet de conseil
-Equipe qualité de l'entreprise
-Autre : ......
3) Exportation vers le marché nord-américain (OUI/NON):
-J'exporte déjà
-J'envisage d'exporter
-Non concerné
4) Préféreriez-vous un outil (OUI/NON):
- téléchargeable comprenant un fichier Excel
automatisé
- exploitable directement sur le web (via un navigateur web)
retour sommaire