Master Qualité - Communication
publique des résultats d'un projet d'intégration Master Qualité
- UTC - Rue du docteur Schweitzer - CS 60319 - 60203
COMPIEGNE Cedex - France - master-qualite@utc.fr
- Tél : +33 (0)3 44 23 44 23
Avertissement
: Si vous arrivez directement sur cette page, sachez
que ce travail est un rapport d'étudiants et doit être
pris comme tel. Il peut donc comporter des
imperfections ou des imprécisions que le lecteur doit
admettre et donc supporter. Il a été réalisé pendant
la période de formation et constitue avant-tout un
travail de compilation bibliographique, d'initiation
et d'analyse sur des thématiques associées aux
concepts, méthodes, outils et expériences sur les
démarches qualité dans les organisations. Nous ne
faisons aucun usage commercial et la duplication est
libre. Si, malgré nos
précautions, vous avez des raisons de contester ce
droit d'usage, merci de nous
en faire part, nous nous efforcerons d'y
apporter une réponse rapide. L'objectif de la
présentation sur le Web est de permettre l'accès à
l'information et d'augmenter ainsi les échanges
professionnels. En cas d'usage du document, n'oubliez
pas de le citer comme source bibliographique.
Bonne lecture...
Référence
bibliographique à rappeler pour tout usage : Le Marquage CE ? C’est
Facile !, Bruno DE TODARO, Ronan DROAL, Anais GIANOLIO,
Nicolas MASSING, Nicolas TREHOUR. Université
de Technologie de Compiègne, Master Qualité et
Performance dans les Organisations (QPO) et Master
Technologies et Territoires de Santé (TTS),
Mémoire d'Intelligence Méthodologique du projet
d'intégration, janvier 2016, www.utc.fr/master-qualite,
puis "Travaux", "Qualité-Management", réf n° 335, https://doi.org/10.34746/dy33-hw21
RESUME
Le marquage CE ? C’est
facile !
Les fabricants de Dispositifs
médicaux (DM) sont parfois rebutés par la longueur de la
procédure d’obtention du marquage CE et l’investissement
que cela représente, notamment en temps pour s’approprier
les directives européennes.
Afin de les éclairer sur les démarches
et la chronologie à suivre pour obtenir ce sésame. Un
guide pratique simplifié a été élaboré, basé sur la
directive 93/42/CEE. Dans un second temps, un didacticiel
open source, utilisant l’interface Scenarii Process a
aussi été développé. Il est mis gratuitement à disposition
et permet de répondre aux interrogations des fabricants
néophytes de DM, de manière ludique.
Mots clés : Marquage CE, Directive 93/42/CEE,
Dispositif médicaux
ABSTRACT
CE Labelling? Such an easy thing to
get!
Medical devices (MEDDEV)
manufacturers can sometimes be discouraged by the
investment required to obtain the CE Label and the time
necessary to learn the EU directive.
The requirements of the EU directive
have been organized, in a chronological order, and made
clear. Therefore, general guidelines have been made and
based on the 93/42/CEE directive. Then,
a tutorial using a, Scenarii Process platform have been
developed. It’s open access and able to answer questions
of beginners manufacturers.
Keywords: CE Mark, 93/42/CEE directive, Medical
device
Nous souhaitons remercier les personnes qui
ont contribuées de près ou de loin à l’élaboration de ce rapport.
Nous remercions l’équipe pédagogique du Master
Qualité et Performance dans les Organisations de l’Université de
Technologie de Compiègne et notamment, M.Farges et M.Istrate qui
nous a suivi au cours de nos travaux.
Nous remercions également, M. Rebiai, CEO de la
société Stream Vision qui nous a fourni une vision concrète et
applicative du projet en nous permettant de nous baser sur le
développement de son dispositif médical.
Depuis 1990, une réglementation a été mise en
place concernant la mise sur le marché des dispositifs médicaux
(DM) en Europe. Il existe plusieurs directives telles que la
directive 90/375/CEE pour les DM implantables Actifs, la directive
93/42/CEE pour les DM « standards », et la directive
98/79/CEE pour les DM de diagnostic in vitro.
Le marquage CE doit être apposé sur un DM afin
de pouvoir être commercialisable au sein de l’Union Européenne. Le
fabricant doit dans ce cadre, soumettre son dispositif à une
procédure d’évaluation de conformité aux exigences le concernant,
et qui sont décrites dans la directive. Celle-ci dépend de la
classe du Dispositif et pour les classes qui présentent les usages
les plus sensibles, elle doit être réalisée, par un organisme
notifié.
Ce travail a pour but d’accompagner les
entreprises de type PME/TPE, dans leurs démarches d’obtention du
marquage CE, concernant un produit innovant.
Après avoir dressé le contexte et les enjeux
que représentent l’apposition de ce sigle sur un DM, l’étude, la
synthèse de la directive européenne 93/42/CEE et les différentes
étapes à réaliser par le fabricant sont exposés. Une interface
d’hypernavigation sera également présentée. Celle-ci a pour but
d’établir un guide interactif et ergonomique de manière à ce que
l’utilisateur appréhende différemment la démarche, face au nombre
important d’informations contenues dans la directive.
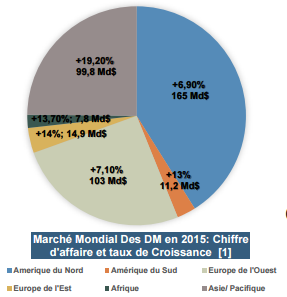
Figure 1 :
Marché Mondial des DM, chiffres 2015 [source : auteurs,
d'après [1]].
En Europe, le marché des
dispositifs médicaux pèse plus de 100 milliards d’euros [2], contre 19 milliards en France [3].
C’est un marché où les innovations technologiques évoluent très
rapidement, avec un nombre de dépôts de brevets par an en nette
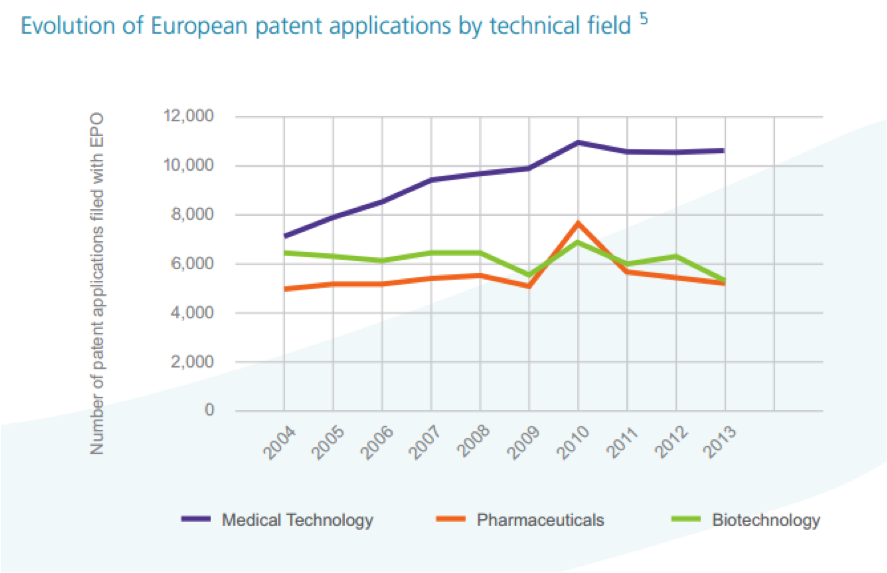
augmentation [Figure2] :
Figure 2:
Nombre de dépôts de Brevets par année en UE [2].
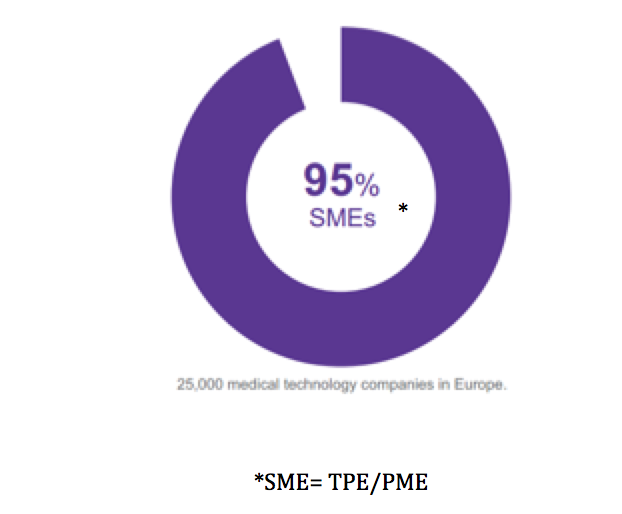
Le tissu industriel des
«medical technologies » dans l’Union européenne compte environ 25000
compagnies dont 95% sont des TPE/PME ou « start up », or le
développement de ces technologies nécessite parfois de lourds
investissements [Figure 2]. Elles investissent en moyenne 6.5% de
leur chiffre d’affaire en recherche et développement (R&D) [2].
Figure 3:
Part des entreprises développant des DMs en UE[2].
Les dispositifs médicaux prennent une place
de plus en plus grande dans le parcours de santé des patients
qui vivent de plus en plus longtemps, avec des pathologies
chroniques. Le marché est donc toujours en forte croissance et
la concurrence est rude.
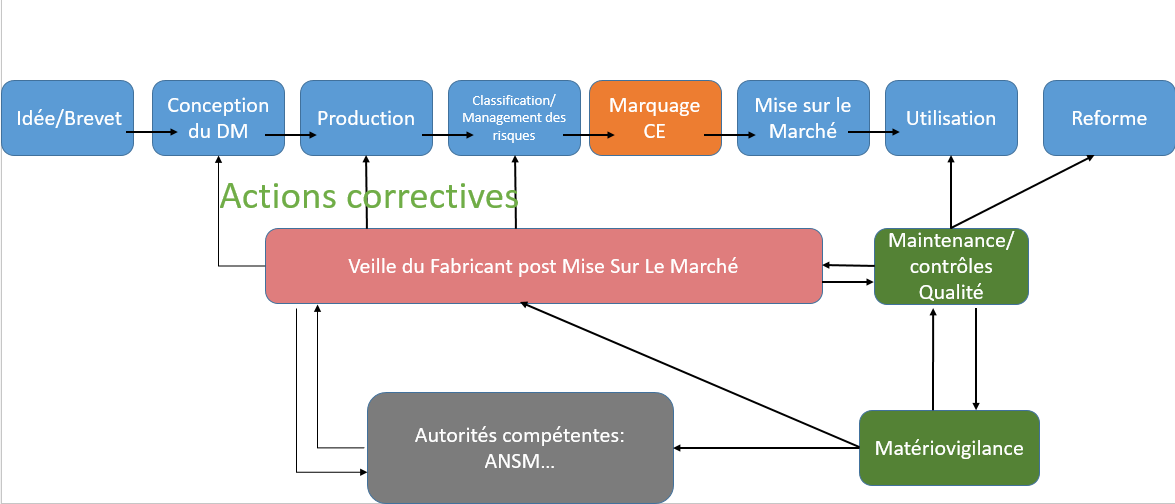
Les
dispositifs médicaux sont utilisés à des fins médicales. Ils doivent
par conséquent être sans risque lors de leur utilisation sur des
patients.
La première preuve de
fiabilité pour ces dispositifs à usage médical, est la conformité
aux exigences européennes du marquage CE. Le fabricant se doit
également, de veiller à la sécurité des utilisateurs et engage sa
responsabilité quant à la fiabilité de son produit, tout au long du
parcours de vie de son dispositif. Il va pour cela interagir avec
différents acteurs qui sont, les exploitants (centres de santé…) et
les sociétés de régulation (ANSM, Organismes notifiés), [Figure 4].
Figure
4: Veille du fabricant et cycle de vie du
DM [source : auteurs].
Le
marquage CE est apparu dans les années 1990, après l’ouverture de
l’espace Schengen qui permet la libre circulation des marchandises
au sein de l’espace européen. Il fallait donc harmoniser les
exigences de sécurité des Dispositifs médicaux pour protéger la
population de produits venant de pays moins exigeants en matière de
sécurité des produits.
Le marquage CE est donc
depuis, obligatoire pour pouvoir mettre sur le marché un dispositif
médical, au sein de l’UE [4]. Il s’agit de ce
fait, d’une phase importante du cycle de vie de ce produit car, plus
le marquage CE est obtenue rapidement et plus vite, plus les petites
entreprises auront la possibilité d’avoir un retour sur
investissement. Cela leur permet aussi de devancer la concurrence
lorsque l’innovation n’est pas protégée par un brevet, et notamment
lors de l’arrivée en Europe des produits chinois et d’occasion.
Le marquage CE d’un DM est obtenu
si le produit et l’entreprise répondent aux exigences décrites dans
la directive européenne qui lui correspond. Il
existe plusieurs directives telles que la directive 90/375/CEE
pour les DM implantables Actifs, la directive 93/42/CEE pour les
DM « standards », et la directive 98/79/CEE pour les DM
de diagnostic in vitro.
Seule la directive UE 93/42/CEE modifiée par la
directive 2007/47/CEE qui s’applique à un plus grand nombre de DM
est étudiée ici et permettra d’aborder la majorité des points
essentiels, pour mener une démarche d’obtention du marquage CE. La
démarche reste en fait identique mais les exigences varient quelque
peu selon la directive.
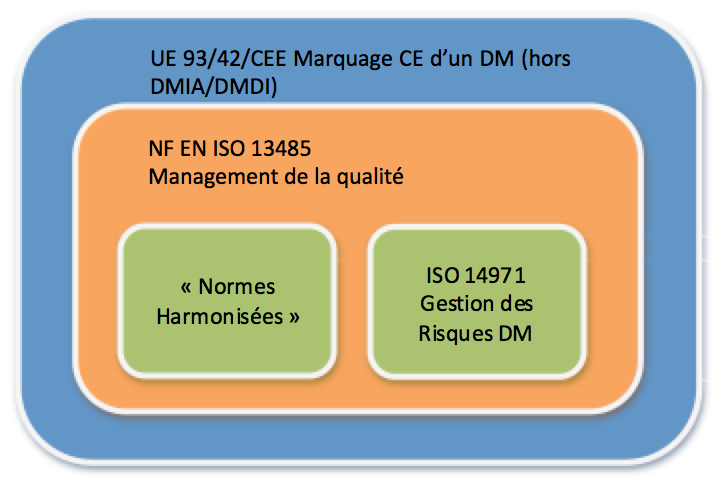
Un certain nombre de normes
dites « harmonisées » à l’ensemble de l’UE, comme décrit
[Figure 5], sont des normes réellement utiles à la conception du
produit. Elles permettent en effet, d’obtenir une présomption de
conformité aux exigences essentielles de la directive. La
recommandation est donc de les appliquer dès les premières phases de
conception du DM. Obtenir les certifications ISO 14971 et NF EN ISO
13485 sont également des facteurs de succès [5].
D’autres normes ISO sont aussi applicables, comme par exemple pour
les DM connectés : l’ISO 62304 et l’ISO 27799. Une liste
exhaustive des normes utiles sera fournie dans ce guide (Chapitre
2).
La
démarche d’obtention du marquage CE est souvent vue comme une
contrainte par les fabricants et d’autant plus s’il s’agit d’une
première. Après un long cheminement, depuis l’émergence de l’idée
jusqu’aux premiers prototypes, qui sont coûteux, cette contrainte a
tendance à rebuter voire décourager les « start up »
biomédicales (très nombreuses sur ce secteur). En effet, cette étape
demande un investissement supplémentaire, et tout particulièrement
en terme de temps, au moment où l’entreprise aurait davantage besoin
d’obtenir des retours sur investissement pour pouvoir se développer.
Le fabricant n’a de plus pas nécessairement, de connaissances en
matière de management de la qualité et appréhender la directive
93/42/CEE lui semble donc une montagne de plus à gravir.
L’étude du contexte et des
enjeux pour les PME ont ainsi amené à se demander : Comment faciliter la démarche
d’obtention du marquage CE des DM, pour une TPE/PME ?
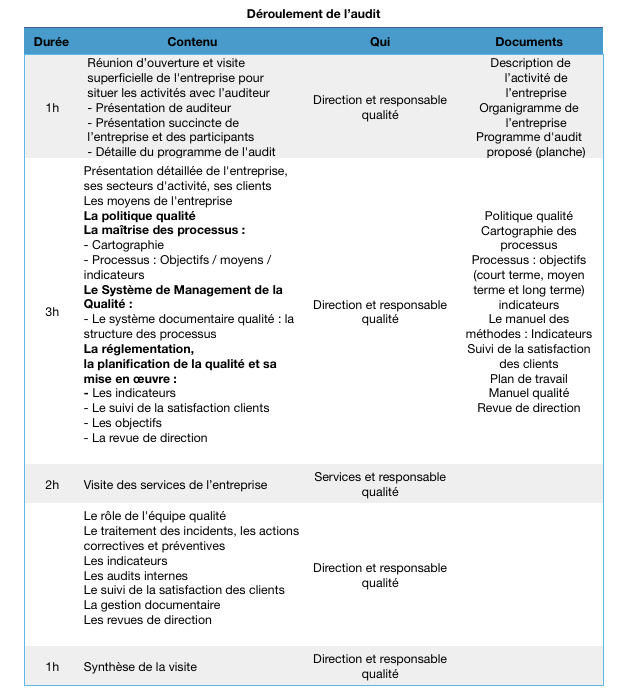
Les étapes d’aide au Marquage CE vont être
exposées de manière chronologique afin d’éclairer les fabricants dans l’obtention du fameux
sésame. Un guide sur le marquage CE des DM a donc pu être élaboré
dans le chapitre 2. Enfin, une interface d’hypernavigation, sera
développé et mis à disposition. Il sera présenté pour venir en
aide à ces entreprises, et leur permettre d’anticiper, dès les
premières phases de conception de leur dispositif, les processus à
piloter, afin que leurs produits répondent aux exigences de
sécurité en vigueur.
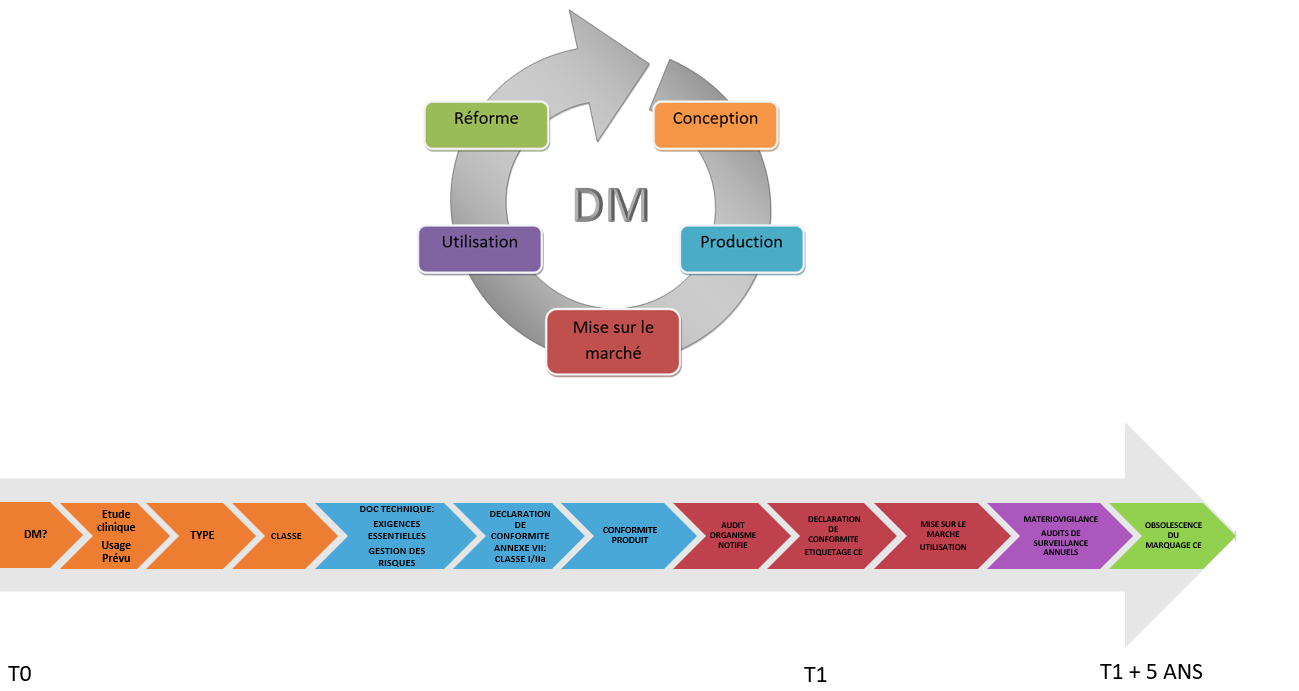
Chapitre 2 : La Démarche d’obtention d’un
Marquage CE des DM : Guide pratique
Pour coller aux exigences
imposées par la directive 93/42/CEE, il est impératif pour le
fabricant de maitriser la qualité de son dispositif sur l’ensemble
des phases de son cycle de vie. Le guide et l’interface
d’hypernavigation s’appuieront donc sur le cycle suivant [Figure 6],
pour établir la chronologie des étapes à réaliser au cours de cette
démarche.
Figure 6 : Cycle de vie du DM et Chronologie
du marquage CE [source : auteurs].
La réglementation de la
filière des dispositifs médicaux concerne trois acteurs :
les fabricants et leurs sous-traitants,
les distributeurs ou mandataires,
les utilisateurs (praticiens, infirmiers, médecins,
chirurgiens, biomédicaux, etc…)
Il existe au moins trois points communs essentiels entre les pays
producteurs de dispositifs médicaux au sujet de la réglementation
:
La Sécurité : déterminer le risque potentiel
lié à l’utilisation du DM; assurer la conformité et la
sûreté du produit de façon durable grâce à un système
d’assurance qualité; une surveillance capable de détecter
les problèmes associés à l’exploitation du DM
(matériovigilance)
Le Fonctionnement : vérifier les performances
techniques du DM : la technologie est t-elle assez
fiable ? Quel va être son cycle de vie du fait de
l’évolution technologique ou de son l’obsolescence ?
L’Utilité : Présente-il assez d’études cliniques
positives pour être jugé utile ? Après on examine l’efficacité
clinique de l’appareil, c’est à dire le rapport
bénéfice/risque. Et enfin, l’efficience du DM :
combien de patients pourraient bénéficier du DM ? Pour quel
type d’opération est-il recommandé ?
NB : Les démarches menant à la mise sur le marché d’un
dispositif médical conformément aux directives 93/42/CEE,
90/385/CEE et 98/78/CE demande de passer par les étapes
précédemment énoncés qui ont nécessairement abouti à des choix.
Une orientation mal définit aura pour conséquence un arrêt ou un
allongement inévitable des délais et une augmentation des coûts
pour l’obtention du certificat CE de type.[6] [7]
La phase de conception est
une phase critique pour prétendre à obtenir un marquage CE. Il
convient lors de cette phase pour le fabricant, de conditionner,
très en amont du projet, ses choix de conception (techniques
utilisées et matériaux), de manière à ce qu’ils répondent aux
exigences de la directive. Ces exigences essentielles visent à réduire
à leur minimum, les risques encourus par les utilisateurs du
dispositif.
Il est donc impératif de
prendre connaissance de ces exigences, ainsi que des normes
européennes harmonisées, dès le début de la phase de
conception du produit. Ces normes sont utiles à appliquer
car elles font présomption de conformité aux exigences
essentielles au regard de l’organisme notifié.
La toute première question à
se poser lorsque l’on souhaite démarrer un processus d’obtention
d’un marquage CE est : Mon produit est-il bien un dispositif
médical, ou plutôt un produit de bien être ?
Selon les termes employés
dans la directive 93/42/CEE, un DM est spécifié dans
l’article premier, 2.a comme :
« Tout
instrument, appareil, équipement, matière ou autre article,
utilisé seul ou en association, y compris le logiciel nécessaire
pour le bon fonctionnement de celui-ci, destiné par le fabricant à
être utilisé chez l’homme à des fins :
De
diagnostic, de prévention, de contrôle, de traitement ou
d’atténuation d’une maladie,
De
diagnostic, de contrôle, de traitement, d’atténuation ou de
compensation d’une blessure ou d’un handicap,
D’étude
ou de remplacement ou modification de l’anatomie ou d’un
processus physiologique
De maîtrise de la conception,
Et dont l’action principale
voulue dans ou sur le corps humain n’est pas obtenue par des
moyens pharmacologiques ou immunologiques ni par métabolisme, mais
dont la fonction peut être assisté par de tels moyens. »
La distinction entre DM et produit de bien être peut parfois
s’avérer subtile et réside alors dans l’usage prévu (la
destination du produit). Par exemple, un cardio-fréquencemètre
utilisé via une montre à visée sportive n’est pas un DM (Cf
Chapitre 2 I.5.).
Le fabricant a ensuite
besoin de savoir à quelle famille appartient le DM. Cette famille
conditionne, la directive européenne à appliquer dans le cadre du
marquage CE et la classification du produit. Les principales
familles qui sont décrites dans les directives européennes sont :
Les dispositifs destinés au diagnostic in vitro (98/79/CEE)
Les dispositifs implantables actifs (90/385/CEE)
Les dispositifs médicaux « classiques » (93/42/CEE)
Les médicaments (65/65/CEE) et médicaments dérivés du sang
(89/381/CEE)
Les produits cosmétiques (76/768/CEE)
Les équipements de protection individuelle (89/686/CEE)
Les autres produits non couverts par la directive 93/42/CEE
:
Sang, plasma, produits
sanguins, cellules sanguines d’origine humaine ou aux
dispositifs qui en contiennent
Organes, tissus,
cellules d’origine humaine et produits dérivés
Organes, tissus,
cellules d’origine animale et produits dérivés (sauf si
utilisés dans la fabrication à l’état non viable)
On entend par : dispositifs médicaux
« classiques », les dispositifs décrits dans le
chapitre 2, I.1.
Un dispositif médical
implantable actif est défini dans la directive 90/385/CEE
(article 1, 2.c) comme : « Tout dispositif médical actif
qui est conçu pour être implanté en totalité ou en partie, par
intervention chirurgicale ou médicale, dans le corps humain ou,
par une intervention médicale, dans un orifice naturel et qui est
destiné à rester après l’intervention. »
Un dispositif médical de
diagnostic in vitro est défini dans la directive 93/42/CEE
(article 1, 2.c) comme : « Tout dispositif qui consiste
en un réactif, produit réactif, ensemble, instrument, appareil ou
système utilisé seul ou en combinaison, destiné par le fabricant à
être utilisé in vitro dans l’examen des échantillons provenant du
corps humain dans le but de fournir une information concernant des
états physiologiques ou des états de santé ou de maladie ou
d’anomalie congénitale. »
Les risques liés à
l’utilisation du dispositif médical, doivent être analysés puis
maîtrisés en couvrant tout le cycle de vie du DM : de la
conception à la réforme par les exploitants.
La norme ISO EN 14971 : 2012
décrit la gestion des risques liés aux dispositifs médicaux, et
peut être appliquée dans ce cadre.
Le plan de gestion des
risques permet d’anticiper les dangers pour les usagers et donc de
les corriger en imaginant des alternatives. Cela jusqu’à avoir
réduit ces risques à un niveau résiduel acceptable au regard des
bénéfices apportés, et qui garantit : la sécurité des
utilisateurs et des patients avant mise sur le marché. Le
Fabricant tient à jour cette gestion des risques et y intègre tout
nouveau danger lié aux signalements de matériovigilance.
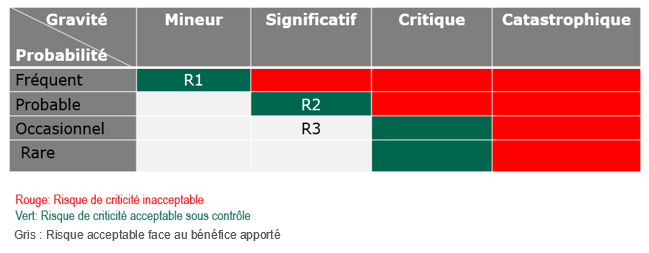
Le Niveau de Risque peut
être estimé comme étant le produit de la probabilité d’apparition
du risque, par la gravité du dommage qu’il est susceptible de
causer (R = P x G).
De
manière qualitative, l’analyse des risques contenue dans le plan
de gestion des risques peut être formalisée avec une matrice
(avant et après mesures correctives) [Figure 7]
Figure 7 : Matrice des risques R
d’un DM [source : auteurs].
Comme vu précédemment dans
le chapitre 2, I.1. Il est impératif pour le fabricant, de fixer
la destination de son DM, assez tôt lors de ses démarches, car
cela va conditionner la classe de dangerosité de celui-ci. Cette
classe conditionnera ensuite elle-même, les exigences en terme
d’assurance de la qualité, qu’il faudra appliquer à ce DM.
Pour définir cet usage prévu
et les conditions normales d’utilisation, le fabricant aura besoin
de s’appuyer sur une étude clinique, afin de vérifier la
faisabilité de son projet et la concordance de son prototype avec
la destination envisagée du DM, et la réponse adéquate au besoin
clinique.
La classification des DM
« classiques » selon la directive 93/42/CEE est
exposée dans cette partie:
La destination du dispositif
médical permettra de distinguer dans un premier temps si le DM en
question est de type :
Non
Invasif
Invasif
chirurgical
Invasif
non chirurgical
Actif
Cas
particuliers
De ce type, en découlera les
règles de classification à appliquer.
En Europe, définir la classe
du DM est nécessaire pour le fabricant. Cela va lui permettre de
définir les contraintes pour établir la conformité du produit aux
exigences réglementaires.
Les dispositifs sont répartis en classe I, classe IIa, classe IIb
et classe III. Elle est directement liée à la dangerosité du
produit. C'est-à-dire, plus le risque sera élevé pour le patient
ou l’utilisateur et plus la classe sera importante.
Afin de pouvoir la déterminer, le fabricant s’appuie sur les 18
règles de l’annexe IX de la directive 93/42/CEE. (Voir glossaire).
Classe I : de faible risque. Peut faire l’objet de données
de vigilance conduisant à des mesures correctives. On y
retrouve par exemple les lits médicalisés, les champs
opératoires, les stéthoscopes…
Classe
IIa : Risque potentiel modéré. Notamment pour le contact
avec les plaies ou les fonctions diagnostiques. On y
retrouve les aiguilles pour seringues, les thermomètres…
Classe
IIb : risque potentiel élevé. Dispositifs radiogène ou
encore les implants passifs. On y retrouve les machines de
dialyse, des oxymètres, les préservatifs masculins…
Classe III :
risques très élevé. Concerne les DM en interaction avec le
système cardio-circulatoire central ou le système nerveux
central. On y retrouve les cathéters destinés au cœur, les
pinces souples de biopsie, des pompes cardiaques…
On peut définir la classe du dispositif grâce à l’outil scenarii
process concernant le marquage CE. [8]
Les études cliniques d’un
dispositif médical se font surtout avant la mise le marché et
également après, au cours de sa commercialisation.
L’annexe I de la directive
93/42/CEE relative aux dispositifs médicaux, mentionne l’exigence
suivante : « la démonstration de la conformité aux
exigences essentielles doit inclure une évaluation clinique
conformément à l’annexe X ».
L’évaluation clinique est
demandée pour tous les dispositifs médicaux excepté ceux de
classe I non stériles.
Suivant l’annexe X de la
directive 93/42/CEE, l’évaluation clinique correspond à
l’analyse des données cliniques d’un dispositif médical
utilisées suivant les indications émissent par l’entreprise. Les
données cliniques sont des informations relatives à la sécurité et
aux performances du dispositif médical dans le cadre d’une
utilisation clinique.
La norme harmonisée NF
EN 14155 relative aux investigations cliniques des
dispositifs médicaux reprend les revendications des Exigences
Essentielles. [9]
L’étude repose sur des
procédures scientifiquement fondées et sur une méthodologie. Les
guides méthodologiques sont les suivants :
MEDDEV
2.7/1 Clinical evaluation: a guide for manufacturers and
notified bodies,
MEDDEV
2.7/4 Guidelines on Clinical investigations: a guide for
manufacturers and notified bodies
MEDDEV
2.12/2 Post market clinical follow-up studies.
Ces guides ont été rédigés
par les Organismes notifiés et les parties prenantes sur la base
de la directive 93/42/CEE. Cette démarche va permettre d’évaluer
la sécurité et la performance du DM sans favoriser de bons
résultats et omettre les résultats moins satisfaisants. Elle doit
permettre un jugement objectif [10].
Démontrer
la sécurité pour chaque indication revendiquée par le
fabriquant,
Démontrer
les performances pour chaque indication revendiquée par le
fabriquant,
Assurer une
analyse critique des données cliniques, [10]
Le fabriquant s’engage sur la pertinence des données cliniques
utilisées pour démontrer la sécurité et les performances du
dispositif médical. Il s’engage :
Sur la
pertinence de ses résultats cliniques
ou
Sur des
résultats critiques à propos de sécurité et de performance,
issus de littérature scientifique validée, ayant un
rapprochement avéré avec les caractéristiques techniques du
DM.
ou
Sur une
évaluation critique à la fois des données cliniques issues
de la littérature et des investigations cliniques réalisées
sur le dispositif.
Ces deux
dernières procédures doivent mettre en évidence :
une
équivalence du DM par rapport au dispositif auquel se
rapportent les données issues de la littérature.
le respect
des exigences essentielles applicables.
En phase de conception, par
un benchmark des dispositifs médicaux et de leurs
caractéristiques, le fabriquant peut commencer à démontrer de
manière rigoureuse l’équivalence de son dispositif avec les
dispositifs déjà présents sur le marché et donc déjà marqués CE.
Mais cette équivalence entre deux appareils identiques est rare.
Un réseau de Centres
d’Investigation Clinique - Innovation Technologique (CIC-IT) (8 en
France) est implanté dans les établissements de soins et de
recherche : ce sont les Centres Hospitaliers Universitaire (CHU).
Il accompagne la recherche et le développement d’entreprises en
faisant le lien entre les chercheurs et les cliniciens selon leurs
domaines de compétence. Sa vocation est de faire émerger des
innovations technologiques médicales. Il participe au
transfert des innovations médicales vers des preuves de concept
et d’évaluation clinique.
Le réseau des CIC-IT
s’adresse aussi bien aux laboratoires, qu’aux TPE/PME et
multinationales. Il favorise davantage l’émergence et la création
de petites entreprises de produits innovants.
Les CIC-IT établissent aussi
des liens étroits avec le Comité de Protection des Personnes (CPP)
et l’Agence Nationale de sécurité du médicament et des produits de
santé (ANSM). Ces contacts directs permettent d’obtenir un avis du
CPP et une réponse de l’ANSM rapide concernant le dossier de
demande d’autorisation d’essai clinique.
L’équipe d’un CIC-IT réunie
des médecins, chercheurs, ingénieurs, informaticiens et
administratifs qui apportent leur savoir-faire scientifique et
organisationnel au cours des études cliniques. Les prestations du
CIC-IT sont les suivantes :
Conseiller d’un point de vu méthodologique, organisationnel,
financier et scientifique, afin d’aider le fabricant à
comprendre les exigences administratives;
Contacter les différents organismes administratifs pour
mettre en œuvre le projet d’étude (CPP, ANSM, DGS (Direction
Générale de la Santé);
Mettre en œuvre l’étude clinique en aidant à la rédaction du
protocole de l’étude, et à la recherche de subventions…
En collaboration avec les cliniciens : recruter des patients
volontaires (malades ou sains) dont les profils sont adaptés à
l’étude (sélection sur une base de données de volontaires);
Apporter une aide technique et organisationnelle en mettant
à disposition des équipements de recherche clinique et en
prenant en charge le projet, dans son ensemble;
Saisir les données ou résultats et les analyser;
Valoriser l’étude clinique en rédigeant des articles
scientifiques et en communicant sur les résultats;
L’ANSM doit être mise au
courant des accidents ou effets néfastes touchant la sécurité des
patients pendant toute la durée de l’étude clinique. L’agence peut
à tout moment suspendre ou interdire la poursuite du projet.
Au sein des CHU, en lien
avec les services hospitaliers, des patients répondant aux
critères d’application du nouveau dispositif médical sont
recrutés. Dans cette phase de conception, l’évaluation du matériel
va permettre également par des retours d’usage, d’améliorer
l’ergonomie du prototype.
Afin
d’obtenir le marquage CE du dispositif, le fabricant doit rédiger
un dossier appelé « documentation technique ». Le dossier doit
comporter les éléments suivants :
l’identification
du
fabricant ;
description
du dispositif, variantes et accessoires ;
destination
et classification ;
plans,
schémas et autres données de construction ;
description
du processus de fabrication et des contrôles effectués (y
compris méthode de stérilisation et sa validation, si
applicable) ;
Étiquetage;
Notice
d'utilisation
;
démonstration
de la conformité aux exigences essentielles
applicables ;
liste
des normes appliquées
analyse
des risques ;
données
cliniques ;
La documentation technique est tenue à la
disposition des autorités compétentes pour tout contrôle ou
inspection qu'elles jugeraient nécessaire. Elle est aussi examinée
par l’organisme notifié pendant l’audit du fabricant.
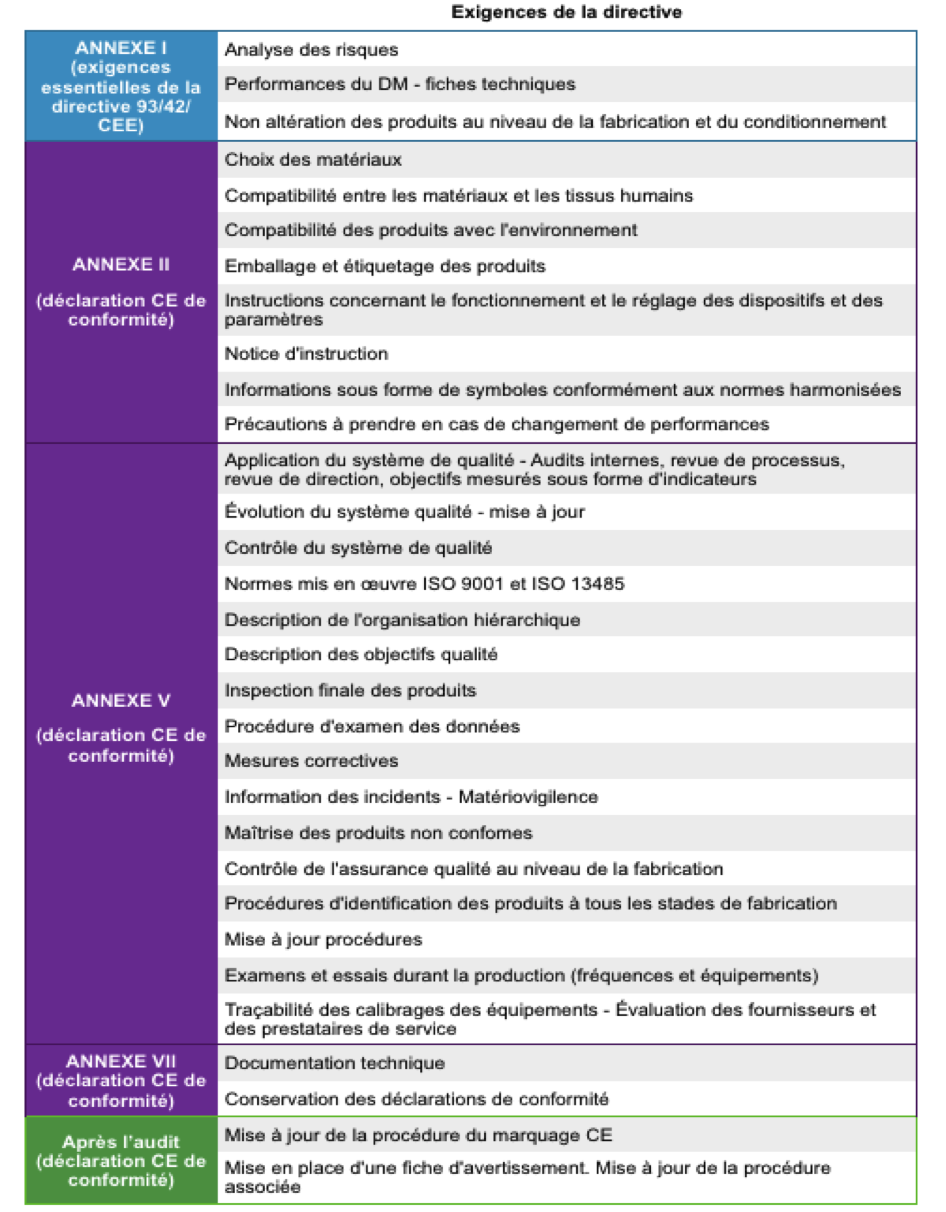
Les exigences
essentielles :
La liste des exigences
essentielles se trouve en annexe de la directive 93/42/CEE. Le
dispositif doit répondre à ces « exigences essentielles » afin de
pouvoir être mis sur le marché. Il appartient au fabricant de
démontrer qu’il a une réponse appropriée pour chaque risque
applicable à son dispositif. Le test du taux de conformité aux
exigences essentielles via l’outil d’autodiagnostic Excel joint à
ce guide est possible.
Les normes harmonisées :
La conformité aux normes
harmonisée n’est pas obligatoire mais ces normes apportent la
présomption de conformité aux exigences essentielles
correspondantes, donc c’est le moyen le plus simple, pour un
fabricant, de démontrer qu’il respecte les exigences essentielles
qui s’appliquent à son produit. Ces normes européennes proposent
des solutions techniques pour répondre aux risques identifiés.
Selon la norme NF EN ISO
14971.
Le fabricant doit apporter la preuve que les risques potentiels
liés à l'utilisation du dispositif médical et trouvant leurs
origines dans tout le cycle de vie (conception, fabrication,
transport, stockage, exploitation et fin de vie du dispositif
médical) sont acceptables au regard du bénéfice apporté au
patient.
Données cliniques :
Le fabricant doit démontrer
que les bénéfices sont supérieurs aux risques et aux effets
indésirables.
Il doit pour cela fournir des « données cliniques », qui peuvent
prendre deux formes principales :
Examen critique de la littérature disponible sur le
dispositif ou sur des produits similaires ;
Ou résultats d’essais cliniques (en particulier lorsqu’il
n’existe pas de littérature disponible, ce qui est le cas pour
les nouvelles technologies ou les applications médicales
innovantes).
La notice d’instruction
permet d'informer les utilisateurs concernant les risques que
l’entreprise n'a pu éliminer par la conception ou la fabrication
de son dispositif. Celle-ci est obligatoire. Le dispositif médical
doit être accompagné d’informations concernant l’utilisation de
celui-ci en tout sécurité.
La notice doit comporter :
Informations
concernant l’étiquetage
Le dispositif
médical doit atteindre les performances qui lui sont
assignés par le fabricant ainsi que les effets secondaires.
Il doit être conçu, fabriqué et conditionné de manière à
remplir une ou plusieurs fonctions assignées par le
fabricant.
Apporter les
indications suffisantes sur les caractéristiques du
dispositif médical afin d’identifier les dispositifs ou
équipements corrects qui peuvent être raccordés à celui-ci
pour fonctionner.
Toutes les
informations nécessaires quant au fonctionnement et à
l’installation du dispositif ainsi que la nature et la
fréquence d’entretien et d’étalonnage afin d’assurer en
permanence le bon fonctionnement et la sécurité du
dispositif.
Les informations
permettant d’éviter certains risques liés à l’implantation
du dispositif.
Informations
relatives aux risques d’interférences réciproques liés à la
présence du DM lors d’investigation ou de traitements
spécifiques.
Les instructions
nécessaires en cas d’endommagement de l’emballage assurant
la stérilité ainsi que la méthode nécessaire pour la
restérilisation.
Les informations
relatives aux méthodes appropriées afin de pouvoir
réutiliser le DM (nettoyage, désinfection, conditionnement,
nombre possible de réutilisations), si celui-ci est
réutilisable. La méthode de restérilisation si celui-ci est
restérilisable.
Les informations
concernant les manipulations ou traitements supplémentaires
nécessaires à la réutilisation du DM (stérilisation,
assemblage final…) exemple : Avant que le dispositif
soit utilisé, il est important de s’assurer que tous les
capteurs sont bien branchés.
Pour un DM
émettant des rayonnements dans un but médical, apporter des
infos sur les rayonnements (type, intensité, nature, et
répartition du rayonnement).
Précautions à
prendre en cas de changement de performances du DM.
Précautions à
prendre en ce qui concerne l’exposition à des champs
magnétiques, des variations de pression, des décharges
électrostatiques, des influences électriques externes, de
l’accélération, des sources thermique d’ignition…
Informations
suffisantes sur les médicaments délivrés par le DM, y
compris les restrictions dans le choix des substances à
administrer.
Précautions à
prendre contre tout risque spécial ou inhabituel lié à
l’élimination du DM.
Les médicaments
incorporés au DM comme partie intégrante de celui-ci. Le
dispositif incorporant une substance peut être considéré
comme un médicament. Voir annexe 1 7.1
Degré de précision
précisé pour les DM de mesurage [11].
Comme vu précédemment, les
risques liés aux dispositifs médicaux influençaient directement la
classe du dispositif médical. Il en est de même pour les exigences
en matière de système d’assurance qualité. C'est-à-dire que plus
la classe du dispositif médical sera importante et plus le système
qualité sera contraignant pour le fabricant. PAS DE PANIQUE !
Tous les cas possibles ainsi que les attentes en matière de
système qualité vont être exposés.
Ainsi, si le produit
fabriqué est un dispositif médical de classe I, une simple
déclaration CE de conformité peut suffire dans la majeure partie
des cas. Cette déclaration CE de conformité est un engagement
écrit par les soins de la société fabricante, cela assure que le
dispositif médical répond aux exigences de la directive.
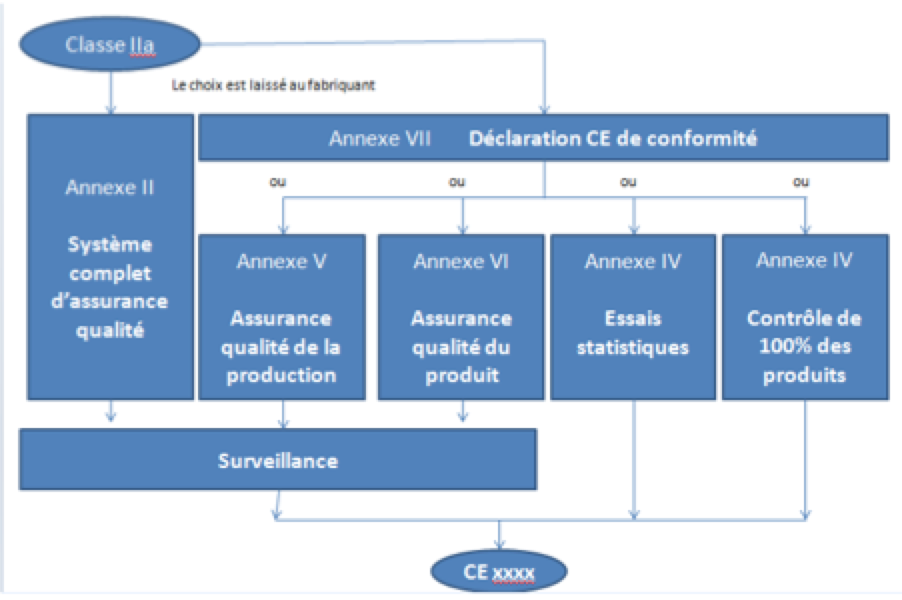
Dans un cas où le dispositif
médical serait de classe IIa, un système qualité doit être mis en
place, comme indiqué dans la figure ci-dessous.
Figure
8 : schéma des choix possibles pour un système qualité
pour un DM de classe IIa [sources : auteurs, d'après [17]].
Pour obtenir le marquage CE
le choix entre la mise en place d’un système complet d’assurance
qualité, OU une déclaration CE de conformité (comme pour la classe
I), est à faire, couplée à l’un des choix suivant :
Assurance qualité de la production
Assurance qualité du produit
Essais statistiques
Contrôle de 100% des produits
Pour un dispositif médical
de classe IIb, les exigences sont sensiblement identiques, mis à
part que la déclaration CE de conformité est remplacée par un
examen CE de type. L’examen CE de type est la procédure par
laquelle un organisme notifié examine et assure que la
documentation associée aux produits est conforme avec ce qui est
réalisé (conception, fabrication, etc)
Pour un dispositif médical
de classe III, le système qualité sera presque le même que pour
une classe IIb. Cependant, des examens et essais seront réalisés
par un organisme notifié, ce qui obligera le fabricant ou
l’exploitant à être d’autant plus avancés dans votre système
qualité. De plus le choix d’assurance qualité du produit n’est
plus disponible pour un dispositif de classe III.
Pour obtenir un complément
d’informations, une interface hypernavigable est disponible et
téléchargeable gratuitement sur cette page web.
Pour toutes les
classes de DM excepté pour la classe I, l’intervention d’un
Organisme Notifié (ON) choisi parmi ceux figurant sur la liste
de la commission européenne, est nécessaire pour évaluer la
conformité prévue par les directives s’appliquant au dispositif
médical. [7][12]
En France il s’agit du G-MED
(organisme de certification dans le milieu médical) qui regroupe
aujourd’hui plus de 800 personnes, désigné par et sous la tutelle
de l’ANSM (Agence Nationale de la Sécurité du Médicament et des
produits de santé).
L’ON est un
organisme certificateur qui délivre les certificats
réglementaires dont ont besoin les fabricants pour mettre leurs
produits sur le marché. L’ON va :
Confirmer le classement du dispositif médical
effectué par le fabricant en tenant compte des règles de
classification définies dans les directives et dans les
guides
En fonction de la classe,
Vérifier la conformité du dispositif médical
(essais, libération des produits fabriqués,...) ;
Vérifier la conformité du système qualité du
fabricant (audit sur site !) ;
Évaluer la documentation technique relative au
dispositif médical, que ce soit systématiquement pour les
produits les plus à risque, ou sur une base
d'échantillonnage pour les produits à risque moyen ;
Évaluer les sous-traitants critiques.
Le fabricant conforme aux
exigences essentielles obtient de la part de l’ON un rapport
d’examen et un certificat d’examen CE de type valable au maximum 5
ans.En cas de projet
engendrant des modifications importantes du produit déjà
examiné, le fabricant doit informer l’ON.
Liste des organismes notifiés dans le domaine des dispositifs
médicaux au 1er décembre 2015 - lien : <http://ec.europa.eu/growth/tools-databases/nando/index.cfm?fuseaction=directive.notifiedbody&sort=country&dir_id=13>
NB : Trois O.N couvrent 60 % des nouveaux DM à savoir le GMed
français, le TÜV product allemand, et en troisième le BSI anglais.
Les audits du système qualité
sont effectués par l’Organisme Notifié annuellement dans le cadre
du cycle de certification du DM. Ils ont pour objectif de vérifier
la documentation et sa mise en œuvre quantifié suivant la classe
du DM et les choix du fabricant. Le système d’assurance qualité
doit constituer un ensemble cohérent de règles, procédures et
pratiques pour contrôler et gérer l’activité du fabricant de DM.
Il se doit d’être conforme aux exigences réglementaires de la
directive applicable au dispositif médical. [9]
Un audit doit être bien préparé. Les procédures, les fiches et les
indicateurs doivent être tous à jour. Il est conseillé de
reconnaître les problèmes argumentés par l’auditeur. L’organisme
notifié doit être informé de la modification du système approuvé
ou des dispositifs déjà examinés.
L’auditeur va examiner toute la documentation
technique et les essais réalisés pour vérifier la conformité aux
exigences essentielles applicables au dispositif médical pour
les phases de conception, de fabrication et finales des DM
concernés.
L’auditeur évalue le système qualité complet
du fabricant pour les DM et s’assure que le système est appliqué
en permanence.
Système d’assurance qualité de la
production :
L’auditeur évalue le système
qualité du fabricant pour la fabrication ainsi que l’inspection
finale des DM concernés et s’assure qu’il est appliqué en
permanence.
Système
d’assurance qualité des produits :
L’auditeur approuve et
surveille le système d’assurance qualité du fabricant pour
l’inspection finale des DM concernés et s’assure qu’il est
appliqué en permanence.
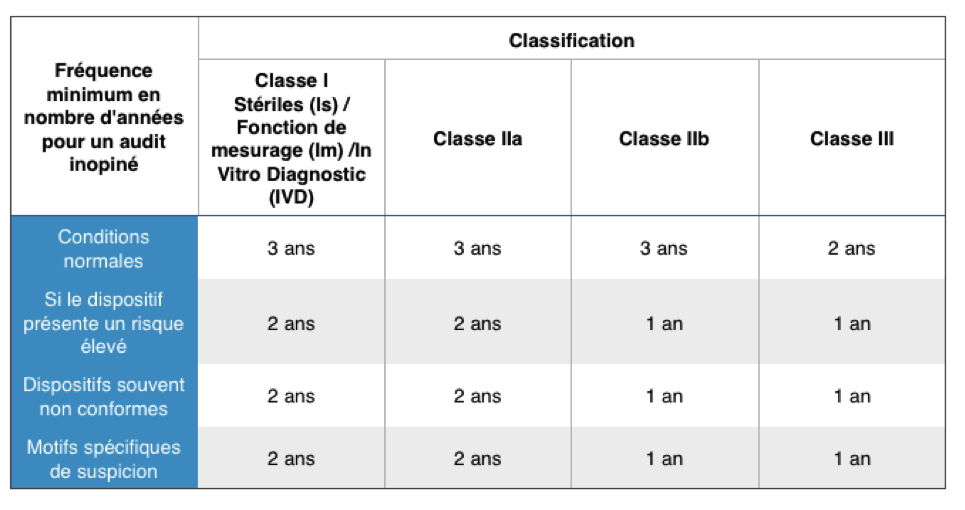
Fréquence des
audits inopinés :
Les audits inopinés sont des
audits supplémentaires. Ils durent au minimum une journée et sont
effectués par au moins 2 auditeurs. Ils sont réalisés chez le
fabriquant ET les sous-traitants ou le fournisseur essentiel. Il
faut partir du principe d’un audit inopiné est réalisé autant de
fois que nécessaire. Dans le cas où la dernière visite s’est
révélée positive, les recommandations de la commission européenne
et des organismes notifiés indiquent les fréquences suivantes:
Une fois le marquage CE
obtenu, il est nécessaire d’y apposer une étiquette CE conforme
aux exigences de la directive 93-42.
Les lettres « CE »
apposées sur les dispositifs médicaux doivent respecter des
proportions prédéfinies par l’annexe XII. Ces différents éléments
apposés ne doivent cependant pas avoir une dimension verticale de
moins de 5 mm, exception faite des dispositifs médicaux de petite
taille, où l’étiquette peut être apposée soit sur l’emballage,
soit sur la notice d’utilisation.
De plus, ce marquage doit
être accompagné du numéro de l’organisme notifié responsable du
marquage (Numéro à 4 chiffres).
Les éléments minimaux qui doivent figurer sur l’étiquette sont les
suivants :
Le nom ou la
raison sociale et l’adresse
du fabricant du dispositif médical, ou le nom du mandataire si
le fabricant ne se retrouve pas dans la communauté européenne.
La dénomination
officielle du produit (ex : Fauteuil de dialyse).
L’identification du
dispositif médical sous une forme précise comme le
numéro de série permettant d’identifier précisément le
dispositif.
Les instructions
particulières d’utilisation tels que les précautions à
prendre, les interdictions ou les différentes mises en gardes
doivent être affichées afin que l’utilisation du dispositif
soit optimale et que l’utilisateur ait conscience des limites
du dispositif et qu’il puisse l’utiliser en toute sécurité.
Le mois etl’année de fabrication
pouvant être incluse dans le numéro de série.
Les conditions
particulières de transport, de stockage et/ou de manutention.
Des cas particuliers exigent une apposition supplémentaire :
Si le dispositif est fait sur mesure, il faut alors le
marquer en toute lettre sur l’étiquette (« Dispositif sur
mesure »).
La mention « STÉRILE » si le dispositif est de
cette catégorie.
Si le dispositif médical possède une date de péremption, la
date limite d’utilisation ne présentant aucun risque pour la
sécurité de l’utilisateur doit être affiché.
La notion précisant que le dispositif est à usage unique le
cas échéant.
S’il s’agit d’un dispositif pour la recherche clinique,
l’apposition « Exclusivement pour investigations
cliniques ».
Les méthodes de stérilisation pour les dispositifs
spécifiques stériles.
S’il existe des substances dérivées du sang humain, elles
doivent être explicitement apposées.
Une évaluation favorable du
système qualité par l’organisme notifié permet au constructeur
d’obtenir un certificat CE de type qui lui permettra d’établir sa
déclaration de conformité nécessaire à la mise sur le marché du
DM.
Il n’y a pas d’autorisation
de mise sur le marché comme pour les médicaments. Le constructeur
prend la responsabilité d’apposer le marquage CE sur le DM après
vérification que les exigences réglementaires soient toutes
accomplies.
Un système de
matériovigilance doit être mis en place, basé sur l’analyse des
risques préalablement établit sur le dispositif médical. Il n’y a
pas d’obligation réglementaire. Cependant la matériovigilance
protège dans une certaine mesure le fabriquant qui déclare une
défaillance dans certaines situations de son dispositif médical.
Il est donc fortement conseillé d’établir une matériovigilance en
adéquation avec l’ANSM pour signaler des événements ou incidents
concernant le dispositif médical.
Une surveillance et un suivi
clinique doivent également être mis en place. Cette démarche va
plus loin que le traitement et le signalement de matériovigilance
et doit permette la collecte d’informations liées à la sécurité et
la performance du dispositif permettant une mise à jour de
l’analyse des risques au cours de sa commercialisation.
La norme ISO 14971 relative
à la gestion des risques des dispositifs médicaux détaille les
exigences réglementaires.
Après la mise sur le marché
du dispositif, l’organisme notifié vérifiera que le fabricant
remplisse toujours les exigences de la directive 93/42/CEE. Pour
cela, il vérifiera annuellement le système d’assurance de la
qualité. En plus d’un audit d’accréditation, il faudra lui
présenter la mise à jour du système de gestion des risques en
fonction des retours d’usage et des déclarations de
matériovigilance. Des audits inopinés peuvent également survenir à
tout moment. Il convient donc de toujours tenir à jours les
preuves documentées et processus de management de la qualité. [13]
Au bout de 5 ans, le
dispositif médical ne pourra plus être vendu. Une nouvelle
procédure de renouvellement du marquage CE sera à faire.
Tout au long de la vie du
dispositif médical, des remarques de clients, des réclamations,
des tests d’évaluation et des suivis de produits sont consignés.
Toutes ces considérations et sources d’informations sont
regroupées dans un dossier permettant la surveillance Post-marché
du dispositif médical. Ce dossier de surveillance post-marché
permet l’amélioration du produit et permet également de procéder à
la gestion des risques et à des actions correctives sur le
dispositif médical.
Cette surveillance
post-marché sera nécessaire lors de l’audit permettant le
renouvellement du marquage CE.
Le renouvellement du
marquage CE sera donc soumis aux mêmes exigences que le marquage
CE de départ, mais avec la fourniture du dossier de post-marché.
Le marquage CE exige pour le
fabricant de dispositif médical de mettre à disposition des pièces
détachées pendant toute la durée du marquage CE. Cette durée étant
de 5 ans.
En outre, il oblige, malgré l’arrêt de production du dispositif,
de pouvoir fournir et remplacer les pièces détachées nécessaires à
sa réparation. Il doit donc, soit avoir un
stock de pièces détachées conséquent, soit pouvoir produire les
pièces détachées afin de pouvoir répondre à cette contrainte.
Une fois ce délai de 5 ans dépassé, le fournisseur ne pourra plus
vendre le dispositif, ou pourra entamer une procédure de
renouvellement du marquage CE.
Deux
règlements européens qui vont remplacer les directives actuelles
sont à l’étude depuis le 09/10/12. Ils concerneront les DM et
les DM de diagnostic in vitro. Ils seront applicables dans les
années à venir (2016-2018) et seront plus précis et plus
lisibles. Les règles à appliquées seront ainsi révisées et
répondront aux manques actuels, notamment au sujet des tissus
humains non viables, ou des produits implantables à but non
médical, par exemple. Des guidelines seront aussi publiées au
sujet de l’évaluation clinique des produits [14].
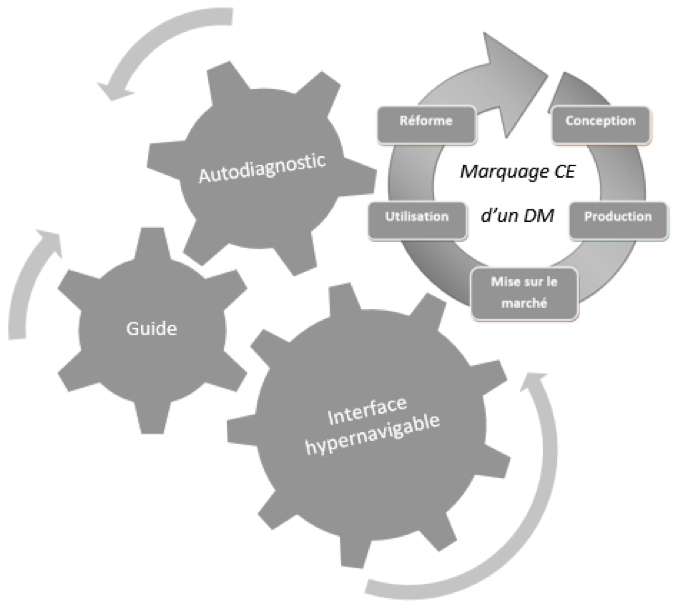
Figure 13:
Outils proposés pour faciliter l’obtention du Marquage
CE d’un Dispositif Médical [sources :
auteurs].
Il est
important de garder à l’esprit que cette solution n’est pas
suffisante pour obtenir le Marquage CE. Elle doit être complétée
par une lecture attentive des directives européennes (93/42/CEE et
2007/47/CEE) et ne remplacera jamais les conseils d’experts.
Un panel d’experts et de professionnels ont néanmoins testés ces
outils. Leurs remarques ont ainsi permis de les rendre plus
opérationnels.
Les différentes étapes du
marquage CE sont fondamentales et doivent répondre aux exigences
essentielles de la directive 93/42/CEE tout au long du cycle de
vie du dispositif médical.
Afin de simplifier la démarche des entreprises
médicales, en particulier pour les TPE et PME, un outil appliqué
au Marquage CE a pu être mis en place via une interface
d’hypernavigation. Il s’agit d’une plateforme qui permet
d’imbriquer des images (type diaporama) et de les connecter les
unes aux autres.
Cet outil reprend tout le cycle de vie d’un
dispositif médical, allant de sa conception jusqu'à sa réforme.
Pour chacune des étapes de son cycle de vie, des exigences doivent
être respectées.
Les démarches menant à la
mise sur le marché d’un dispositif médical conformément aux
directives 93/42/CEE, 90/385/CEE et 98/78/CE demande de passer par
des étapes de sécurité grâce au système d’assurance qualité, de
fonctionnement et d’utilisation qui ont nécessairement abouti à
des choix. Une orientation mal définit aura pour conséquence un
arrêt ou un allongement inévitable des délais et une augmentation
des coûts pour l’obtention du certificat CE de type.
Dans cet outil, les clés et phases à suivre pour simplifier
les démarches d’obtention du marquage CE sont exposées.
L’interface hypernavigable présente pour une TPE ou PME une
solution facile à utiliser dans le but de suivre pas à pas la
démarche du Marquage CE. En sélectionnant les étapes du cycle
de vie du dispositif médical, il sera donc aisé, d’obtenir des
informations sur les différentes exigences imposées.
Les processus sont exposés de façon claire,
sous forme de cartographies et de logigrammes visuels. Cela
constitue une interface intuitive et interactive qui donne un
panorama des étapes clées et des procédures, à appliquer pour
le dispositif médical.
Ce support se démarque des
autres solutions car un livre sur l’obtention du marquage CE, un
dvd, une formation, ou bien d’autres choses auraient pu être
réalisés. Cependant, grâce à cette interface, il est plus simple
de mettre à jour les données en cas d’évolution de la directive.
De plus, un livre prend plus de temps à lire, à comprendre et à
appliquer. Avec l’interface, un guidage tout au long de la
démarche CE est proposée. Une visualisation rapide des critères et
des exigences applicables au DM est possible. L’outil est
disponible gratuitement sur internet et à toute heure. A l’inverse
d’une formation l’accès à cette interface peut se faire selon le
souhait de chacun. L’interface d’hypernavigation débutera par une
représentation graphique du cycle de vie d’un dispositif médical
et offrira la possibilité, par simples clics, d’accéder à des
cartographies interactives explicitant soit directement les
logigrammes de classification des DM, soit l’ensemble des
exigences contenues dans les annexes de la directive 93/42/CEE pas
à pas.
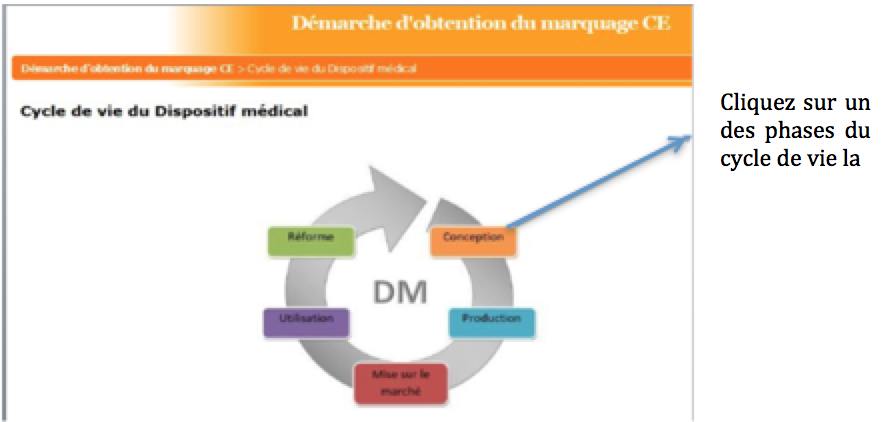
Une fois la phase du cycle sélectionnée, une
navigation dans les menus en sélectionnant les parties qui vous
intéressent sur la frise associée sera possible:
Figure 17: Page de
l'interface [sources : auteurs].
Les exigences essentielles
énoncées dans l’annexe 1 de la directive 93/42/CEE forment le
socle de base, pour assurer la sécurité des utilisateurs du
dispositif médical. Nous avons donc élaboré un outil
d’autodiagnostic, complémentaire de la plateforme Scenarii
développée et qui vous permettra de vérifier si votre dispositif
est conforme à ces exigences.
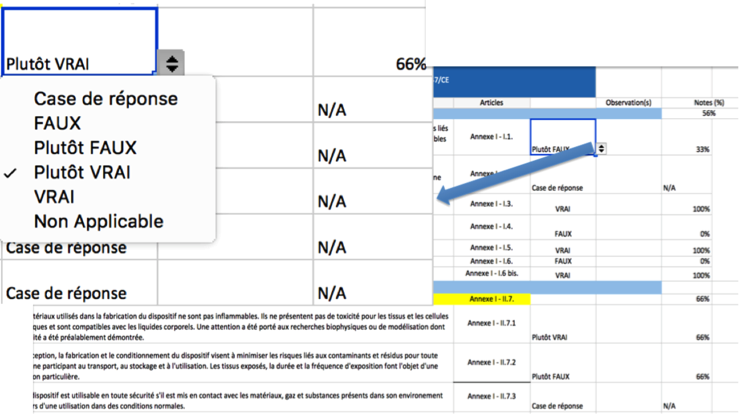
Présentation de l’outil
L’outil est établi sous un
format « tableur Excel », il comprend les 93 critères
de l’annexe 1 qui sont regroupés sous 8 grands thèmes qui
représentent les catégories de dangers (électrique, chimique…).
Vous renseignerez par le biais de « menus
déroulants », le taux de corrélation entre les 93
affirmations et les caractéristiques de votre dispositif afin
d’obtenir un résultat sous forme de représentation graphique.
Vous verrez ainsi rapidement, les points qu’il vous faudra
améliorer, dans la conception de votre produit, afin de pouvoir
obtenir le marquage CE.
L’outil Excel vous est
facilement et gratuitement téléchargeable sur le site du Master
Qualité. La plateforme Excel est connue de tous ce qui vous
permettra une utilisation fluide et rapide grâce aux choix pré
définis (vrai, plutôt vrai, faux…) Vous n’avez plus qu’à cocher
vos réponses et le graphe radar se met à jour automatiquement en
temps réel.
Ce graphique radar vous
permettra de voir en un clin d’œil les axes qui font défaut à
votre appareil, et ainsi pouvoir les corriger pour répondre au
mieux aux exigences essentielles de la directive 93/42/CEE.
Vous pouvez également imprimer vos résultats sous format papier
en A4, qui vous afin de garder des traces de votre
questionnaire.
L’utilisation de l’outil
ne devrait pas vous prendre plus de 35 min mais il faut pour
cela avoir déjà pris connaissance des exigences essentielles de
l’annexe 1 de la directive 93/42/CEE. Nous vous conseillons donc
de prévoir un peu plus de temps pour la première utilisation. Place de la biochimie dans la biologie médicale
Place de la biochimie dans la biologie médicale
Le Marquage CE est une
procédure qui peut paraître très abstraite et lourde pour un
fabricant qui souhaite commercialiser son produit innovant. La
démarche n’est malgré tout pas si compliquée. Par le biais de ce
Mémoire d’Intelligence Méthodologique il a été tenté de créer un
guide simple mais exhaustif des étapes à réaliser pour obtenir ce
marquage.
Il est construit de manière
chronologique et selon le cycle de vie d’un dispositif médical pour
faciliter la compréhension des étapes clés. Ce système semble très
cloisonné mais il faut savoir que certaines étapes sont en réalité
concomitantes. Certaines d’entre elles comme l’étude clinique,
doivent être prévues très en amont du projet et poursuivies sur la
quasi-totalité de la démarche CE. Des informations à ce sujet sont
toutefois exposées dans ce guide.
Ce projet d’aide aux TPE/PME
biomédicales a aussi amené à créer des outils comme une interface
d’hypernavigation et un outil d’autodiagnostic de conformité du DM,
aux exigences essentielles de la directive 93/42/CEE. Ces outils
visent à fournir aux fabricants, une vision plus claire des
processus à piloter lors de leurs démarches.
Il est cependant à noter que,
l’Union européenne tend à faire évoluer le système actuel par des
règlements européens qui éviteraient les différences entre pays,
dues à l’interprétations des directives. Leur application sera
nécessaire dans les années à venir. Une terminologie commune et un
système international commun de matériovigilance permettrait
également d’harmoniser les pratiques et ainsi, d’augmenter la
sécurité des utilisateurs et in fine, de l’ensemble des citoyens
européens.
Définitions utiles pour la classification
(issues de la directive 93/42/CEE modifiée par la
2007/47/CEE) :
La durée :
Temporaire : utilisation en continu
pendant moins de soixante minutes.
Court terme : utilisation en continu
pendant 30 jours au maximum
Long terme : utilisation en continu
pendant plus de 30 jours.
Dispositif invasif :
Invasif : dispositif
qui pénètre partiellement ou entièrement à l’intérieur du corps,
soit par un orifice du corps soit à travers la surface du corps.
Orifice chirurgical : toute ouverture
naturelle du corps, ainsi que la surface externe du globe
oculaire, ou toute ouverture artificielle permanente.
Dispositif invasif de
type chirurgical : dispositif invasif qui
pénètre à l’intérieur du corps, à l’aide ou dans le cadre
d’un acte chirurgical.
Dispositif implantable : destiné à être implanté
en totalité dans le corps humain ou a remplacer une surface
épithéliale ou la surface de l’œil grâce à une intervention
chirurgicale.
Un dispositif implantable
est aussi un dispositif destiné a être partiellement introduit
dans le corps humain par une intervention chirurgicale, et qui est
destiné à rester en place pendant une période d’au moins trente
jours après l’intervention.
Instrument chirurgical
réutilisable : instrument qui est destiné à
réaliser un acte chirurgical comme couper, scier, forer, gratter,
racler, serrer, rétracter ou attacher.
Dispositif actif :
Dispositif médical
actif : tout dispositif ayant besoin d’une source d’énergie
électrique pour fonctionner ou toute autres source d’énergie autre
que celle directement générée par le corps humain les dispositifs
destinés à transmettre de l’énergie ne sont pas considéré comme
dispositif médical actif.
Dispositif actif
thérapeutique :
tout dispositif médical actif, utilisé en combinaison ou non avec
d’autres dispositifs médicaux, permettant de soutenir, modifier,
remplacer ou restaurer des fonctions dans le but le traiter,
soulager une blessure une maladie ou un handicap.
Dispositif actif destiné
au diagnostic : tout dispositif médical actif,
utilisé en combinaison ou non avec d’autres dispositifs médicaux,
permettant de fournir des informations en vue de détecter,
diagnostiquer, contrôler ou traiter des états physiologiques, de
santé, des maladies ou de malformations congénitales.
Système circulatoire
centrale : on entend par « Système circulatoire centrale »
les artères pulmonaires, ascendante, coronaire, carotide commune,
carotide externe, carotide interne, cérébrale, et les veines cave
supérieur, inférieur, pulmonaire.
Système nerveux
central : encéphale, moelle épinière et méninges.
Dispositifs, cas
particuliers :
Tous les dispositifs
incorporant comme partie intégrante une substance qui, si elle
est utilisée séparément, peut être considérée comme un
médicament au sens de l’article 1er de la directive
« 2001/83/CE » et qui est susceptible d’agir sur le
corps par une action accessoire à celle des dispositifs.
Tous les dispositifs
incorporant comme partie intégrante une substance dérivée du
sang humain.
Tous les dispositifs
utilisés pour la contraception ou pour prévenir la
transmission de maladies sexuellement transmissible.
Tous les dispositifs
destinés spécifiquement à désinfecter, nettoyer, rincer ou, le
cas échéant, hydrater des lentilles de contact.
Tous les dispositifs
destinés spécifiquement à désinfecter les DM.
Tous les dispositifs
destinés spécifiquement à enregistrer les images de
radiodiagnostic.
Tous les dispositifs
fabriqués à partir de tissus d’origine animale ou dérivés
rendus non viables sauf, si ces dispositifs sont destinés à
entrer en contact uniquement avec une peau intacte.
[5] Organisme de
certification : Certification de systèmes de management
d’entreprise, LNE/G-MED, , mis à jour le
08/03/2013 ;[consulté en novembre 2015]. Disponible
sur :http://www.gmed.fr/pages/services/certification_smq.asp
Figure 4 : Veille du
fabricant et cycle de vie du DM, Groupe projet, inspiré d’un
cours réalisé par C. JEGOU, « Mise sur le marché d’un
dispositif médical », consulté en Octobre 2015.
Figure 5 : Marquage CE d’un DM, Groupe projet, crée en
octobre 2015.
Figure 6 : Cycle de vie du DM et
Chronologie du marquage CE, Groupe projet, créé en
Novembre 2015.
Figure 7 :Matrice des risques R d’un DM,
Groupe projet, créé en
Novembre 2015.
Figure 8 :Schéma des choix possibles pour
un système qualité pour un DM de classe IIa, Groupe projet, Crée en
Novembre 2015.
Figure 13: Outils
proposés pour faciliter l’obtention du Marquage CE d’un Dispositif
Médical, Groupe projet,
Crée en Janvier 2016. Figure 14 : Page
accueil interface d'hypernavigation source auteurs Figure 15 : Lien
pour accès à cycle de vie du dispositif médical [source auteurs
] Figure
16: Cycle de vie d'un DM [source auteurs ]
Figure 17: Page
de l'interface [source auteurs ]
Figure 18: Page production de l'interface [source auteurs ]
Figure
19: Bouton retour [source auteurs ]
Figure
20: Bouton retour avec accès processus [source auteurs ]